

Reduced PI3K(p110α) induces atrial myopathy, and PI3K-related lipids are dysregulated in athletes with atrial fibrillation
- PMID: 39826614
- PMCID: PMC11978378
- DOI: 10.1016/j.jshs.2025.101023
Reduced PI3K(p110α) induces atrial myopathy, and PI3K-related lipids are dysregulated in athletes with atrial fibrillation
Abstract
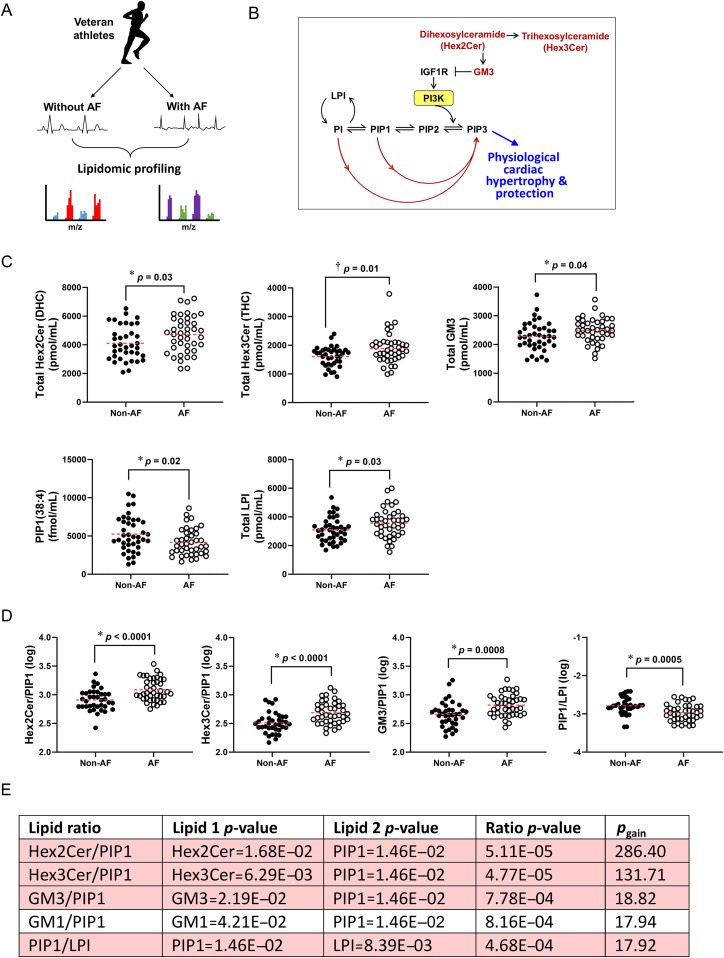
Background: Elucidating mechanisms underlying atrial myopathy, which predisposes individuals to atrial fibrillation (AF), will be critical for preventing/treating AF. In a serendipitous discovery, we identified atrial enlargement, fibrosis, and thrombi in mice with reduced phosphoinositide 3-kinase (PI3K) in cardiomyocytes. PI3K(p110α) is elevated in the heart with exercise and is critical for exercise-induced ventricular enlargement and protection, but the role in the atria was unknown. Physical inactivity and extreme endurance exercise can increase AF risk. Therefore, our objective was to investigate whether too little and/or too much PI3K alone induces cardiac pathology.
Methods: New cardiomyocyte-specific transgenic mice with increased or decreased PI3K(p110α) activity were generated. Multi-omics was conducted in mouse atrial tissue, and lipidomics in human plasma.
Results: Elevated PI3K led to an increase in heart size with preserved/enhanced function. Reduced PI3K led to atrial dysfunction, fibrosis, arrhythmia, increased susceptibility to atrial enlargement and thrombi, and dysregulation of monosialodihexosylganglioside (GM3), a lipid that regulates insulin-like growth factor-1 (IGF1)-PI3K signaling. Proteomic profiling identified distinct signatures and signaling networks across atria with varying degrees of dysfunction, enlargement, and thrombi, including commonalities with the human AF proteome. PI3K-related lipids were dysregulated in plasma from athletes with AF.
Conclusion: PI3K(p110α) is a critical regulator of atrial biology and function in mice. This work provides a proteomic resource of candidates for further validation as potential new drug targets and biomarkers for atrial myopathy. Further investigation of PI3K-related lipids as markers for identifying individuals at risk of AF is warranted. Dysregulation of PI3K may contribute to the association between increased cardiac risk with physical inactivity and extreme endurance exercise.
Keywords: Atrial fibrillation; Atrial myopathy; Exercise; Lipidomics; Proteomics.
Copyright © 2025. Production and hosting by Elsevier B.V.
Conflict of interest statement
Competing interests The authors declare that they have no competing interests.
Figures







References
-
- Chen Y.C., Voskoboinik A., Gerche A.L., Marwick T.H., McMullen J.R. Prevention of pathological atrial remodeling and atrial fibrillation: JACC State-of-the-Art review. J Am Coll Cardiol. 2021;77:2846–2864. - PubMed
-
- Weeks K.L., Gao X., Du X.J., et al. Phosphoinositide 3-kinase p110alpha is a master regulator of exercise-induced cardioprotection and PI3K gene therapy rescues cardiac dysfunction. Circ Heart Fail. 2012;5:523–534. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
