

Construction of the bromodomain-containing protein-associated prognostic model in triple-negative breast cancer
- PMID: 39827145
- PMCID: PMC11742518
- DOI: 10.1186/s12935-025-03648-7
Construction of the bromodomain-containing protein-associated prognostic model in triple-negative breast cancer
Abstract
Background: Bromodomain-containing protein (BRD) play a pivotal role in the development and progression of malignant tumours. This study aims to identify prognostic genes linked to BRD-related genes (BRDRGs) in patients with triple-negative breast cancer (TNBC) and to construct a novel prognostic model.
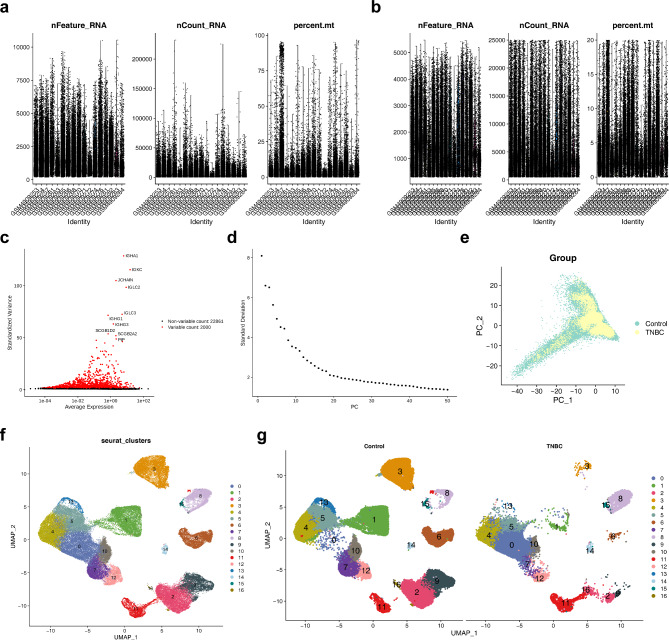
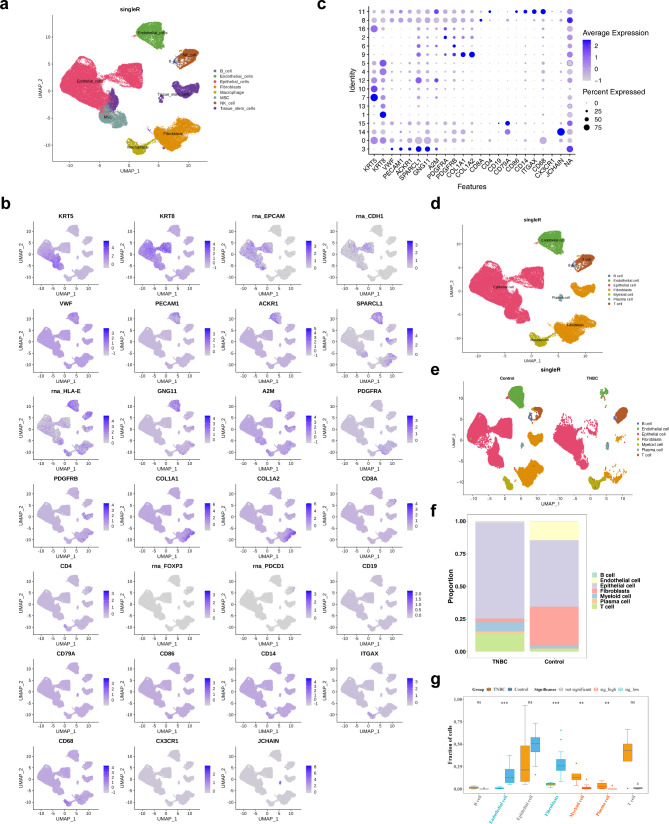
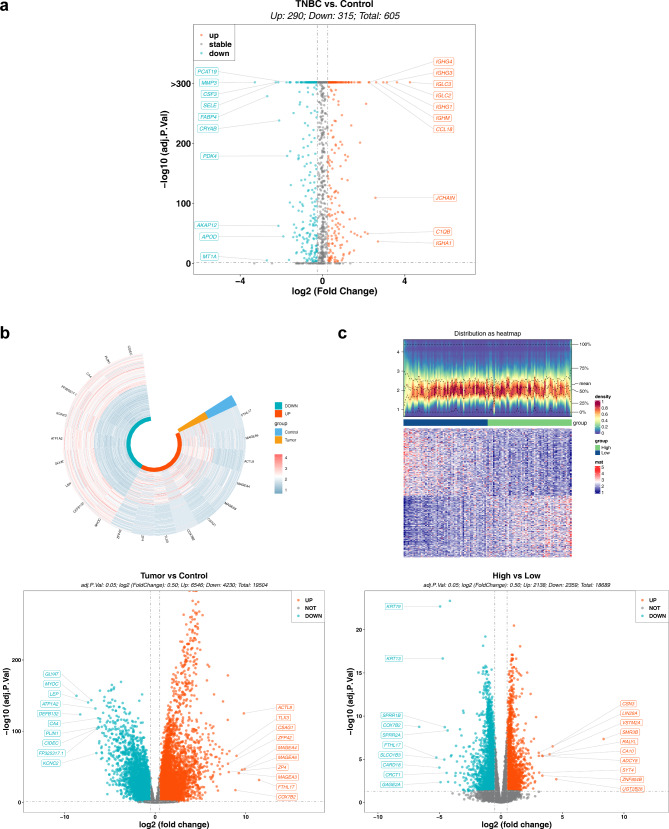
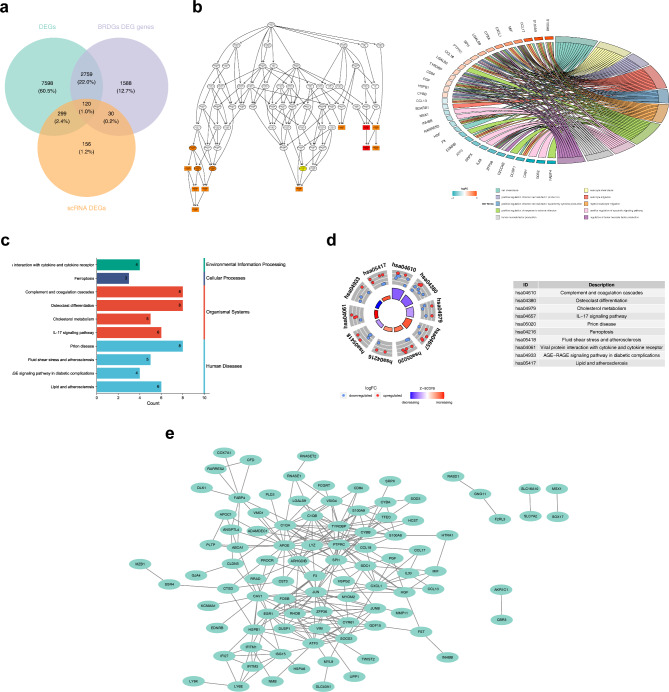
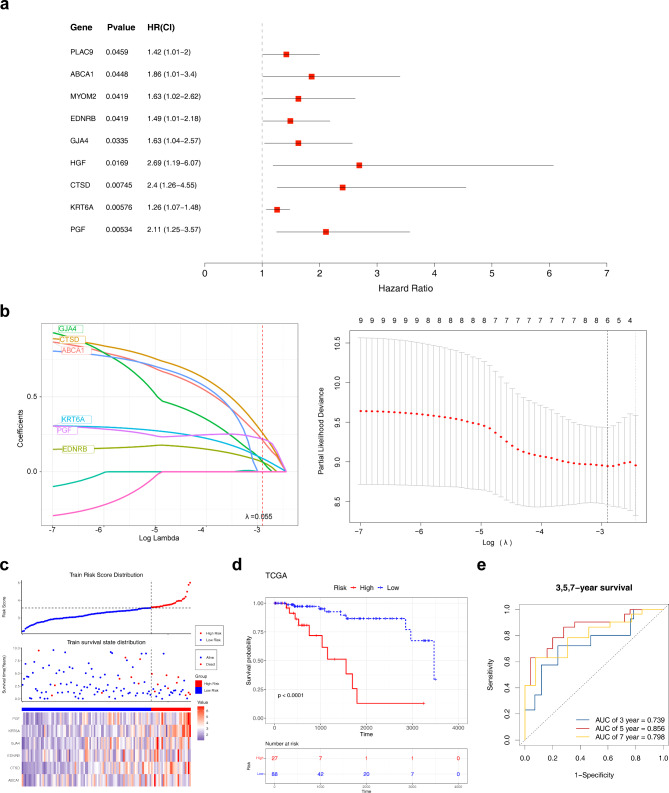
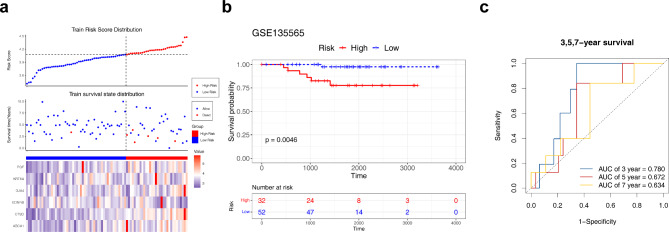
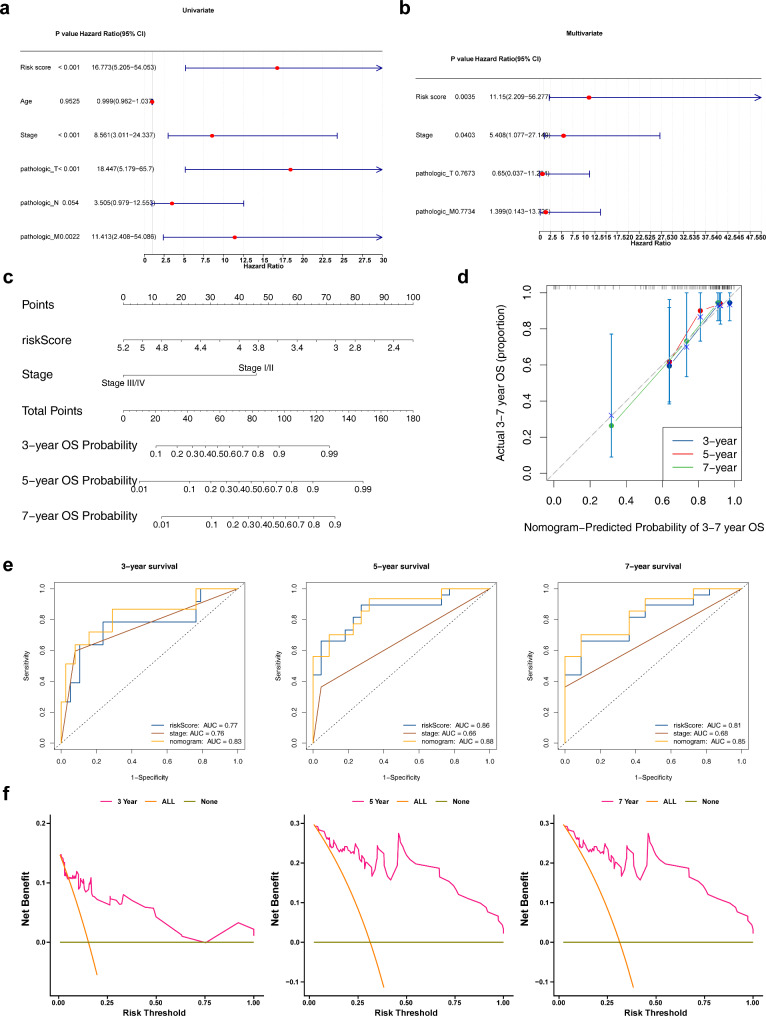
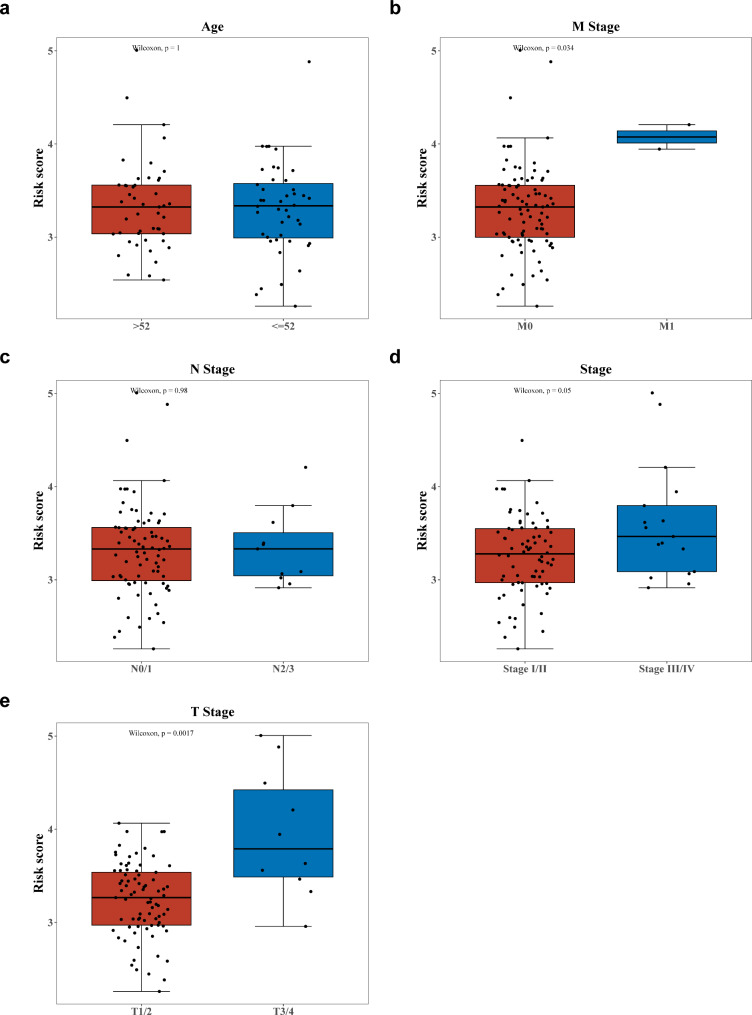
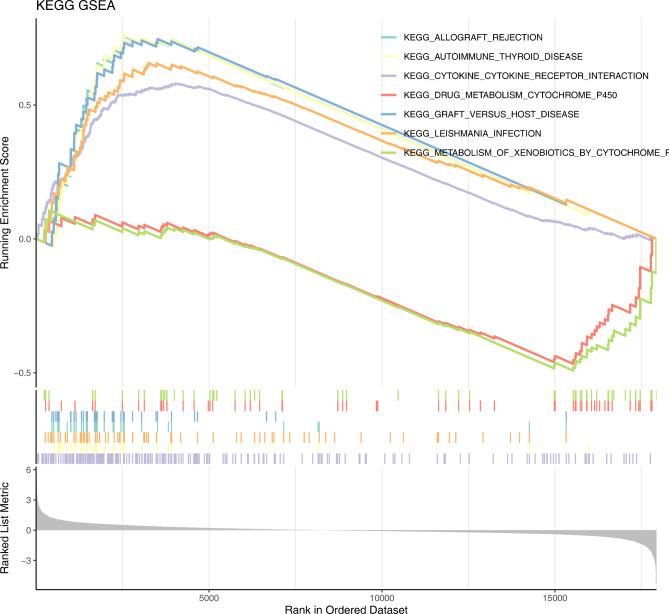
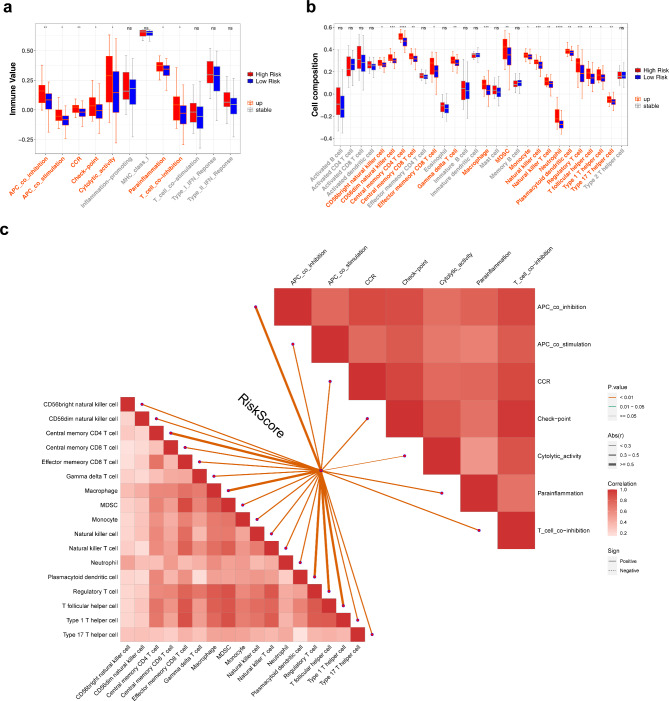
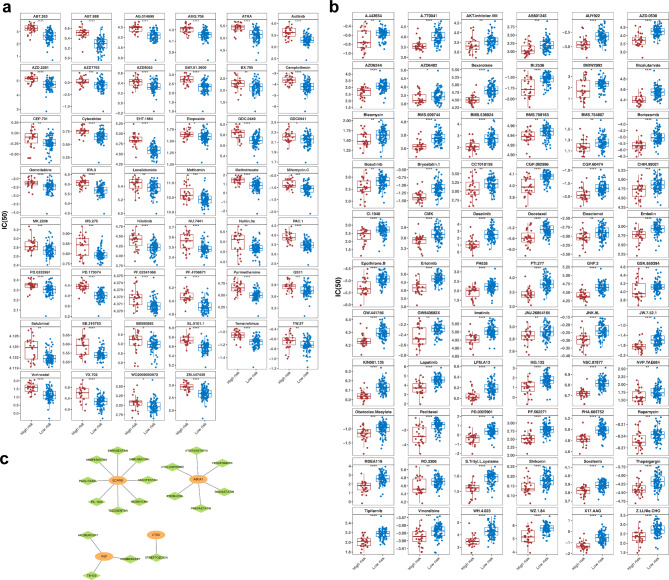
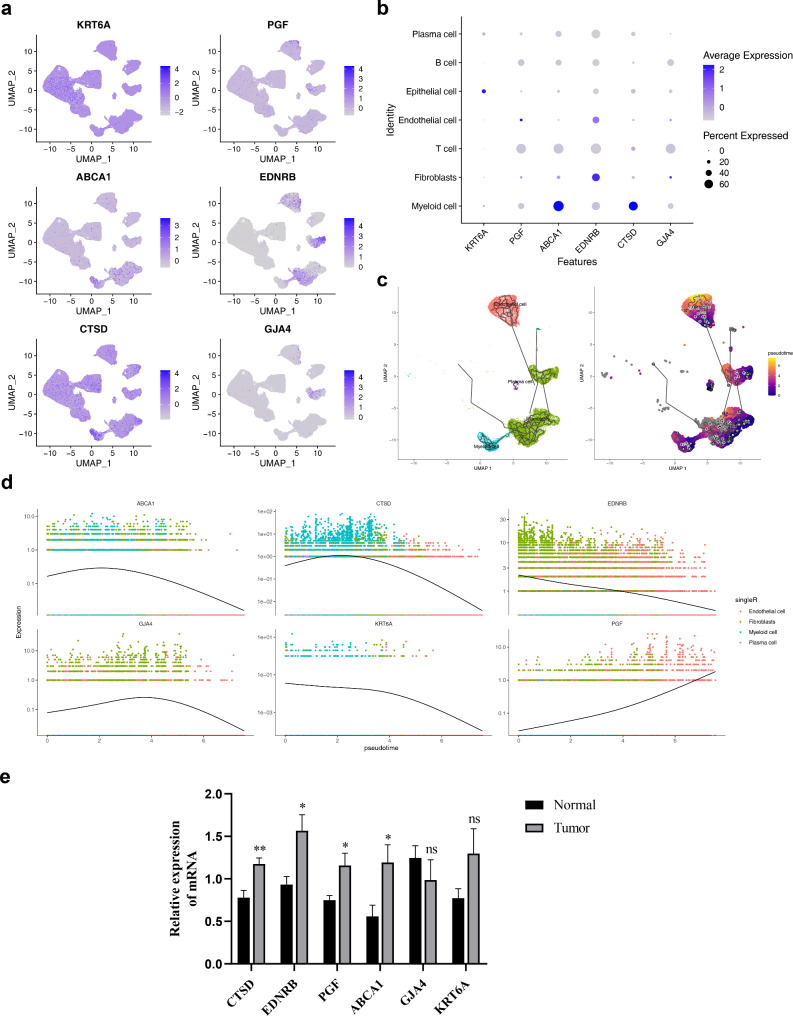
Methods: Data from TCGA-TNBC, GSE135565, and GSE161529 were retrieved from public databases. GSE161529 was used to identify key cell types. The BRDRGs score in TCGA-TNBC was calculated using single-sample Gene Set Enrichment Analysis (ssGSEA). Differential expression analysis was performed to identify differentially expressed genes (DEGs): DEGs1 in key cells, DEGs2 between tumours and controls and DEGs3 in high and low BRDRGs score subgroups in TCGA-TNBC. Differentially expressed BRDRGs (DE-BRDRGs) were determined by overlapping DEGs1, DEGs2 and DEGs3. Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, and protein-protein interaction (PPI) network analysis were conducted to investigate active pathways and molecular interactions. Prognostic genes were selected through univariate Cox regression and least absolute shrinkage and selection operator (LASSO) regression analyses to construct a risk model and calculate risk scores. TNBC samples from TCGA-TNBC were classified into high and low-risk groups based on the median risk score. Additionally, correlations with clinical characteristics, Gene Set Enrichment Analysis (GSEA), immune analysis, and pseudotime analysis were performed.
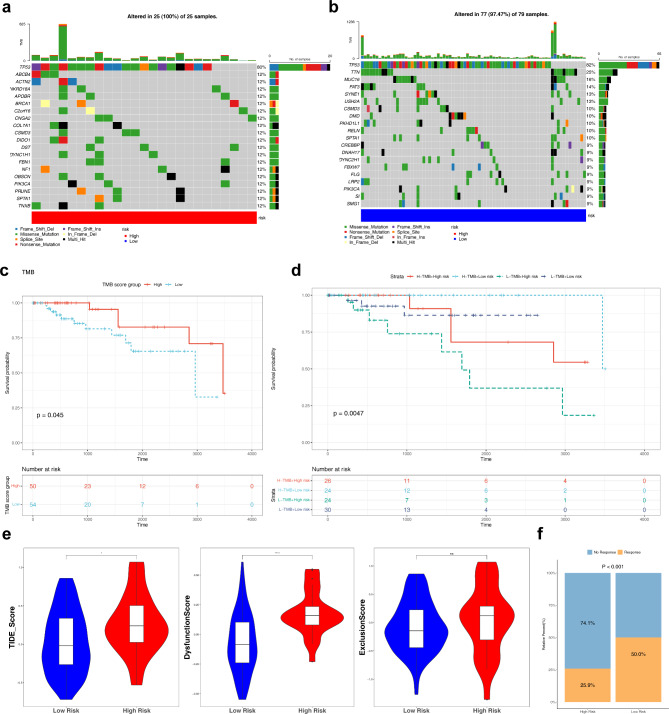
Results: A total of 120 DE-BRDRGs were identified by overlapping 605 DEGs1 from four key cell types, 10,776 DEGs2, and 4,497 DEGs3. GO analysis revealed enriched terms such as 'apoptotic process,' 'immune response,' and 'regulation of the cell cycle,' while 56 KEGG pathways, including the 'MAPK signaling pathway,' were associated with DE-BRDRGs. A risk model comprising six prognostic genes (KRT6A, PGF, ABCA1, EDNRB, CTSD and GJA4) was constructed. A nomogram based on independent prognostic factors was also developed. Immune cell abundance was significantly higher in high-risk group. In both risk groups, TP53 exhibited the highest mutation frequency. The expression of KRT6A, ABCA1, EDNRB, and CTSD went decreased progressively in pseudotime.
Conclusion: A novel prognostic model for TNBC associated with BRDRGs was developed and validated, providing fresh insights into the relationship between BRD and TNBC.
Keywords: Bromodomain-containing protein; Immune microenvironment; Prognosis; Triple-negative breast cancer.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: The study was approved by the Fujian Provincial Hospital Ethics Committee (Approval No: K2021-04-069), and all patients provided written informed consent. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Construction and validation of a prognostic model of angiogenesis-related genes in multiple myeloma.BMC Cancer. 2024 Oct 11;24(1):1269. doi: 10.1186/s12885-024-13024-9. BMC Cancer. 2024. PMID: 39394121 Free PMC article.
-
Screening of DNA Damage Repair Genes Involved in the Prognosis of Triple-Negative Breast Cancer Patients Based on Bioinformatics.Front Genet. 2021 Aug 2;12:721873. doi: 10.3389/fgene.2021.721873. eCollection 2021. Front Genet. 2021. PMID: 34408776 Free PMC article.
-
Integrative analyses of genes associated with oxidative stress and cellular senescence in triple-negative breast cancer.Heliyon. 2024 Jul 16;10(14):e34524. doi: 10.1016/j.heliyon.2024.e34524. eCollection 2024 Jul 30. Heliyon. 2024. PMID: 39130410 Free PMC article.
-
Research and experimental verification on the mechanisms of cellular senescence in triple-negative breast cancer.PeerJ. 2024 Feb 29;12:e16935. doi: 10.7717/peerj.16935. eCollection 2024. PeerJ. 2024. PMID: 38435998 Free PMC article.
-
A novel model associated with tumor microenvironment on predicting prognosis and immunotherapy in triple negative breast cancer.Clin Exp Med. 2023 Nov;23(7):3867-3881. doi: 10.1007/s10238-023-01090-5. Epub 2023 May 23. Clin Exp Med. 2023. PMID: 37219794 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
