

This is a preprint.
Fast simulation of identity-by-descent segments
- PMID: 39829821
- PMCID: PMC11741331
- DOI: 10.1101/2024.12.13.628449
Fast simulation of identity-by-descent segments
Update in
-
Fast simulation of identity-by-descent segments.Bull Math Biol. 2025 May 23;87(7):84. doi: 10.1007/s11538-025-01464-8. Bull Math Biol. 2025. PMID: 40410602 Free PMC article.
Abstract
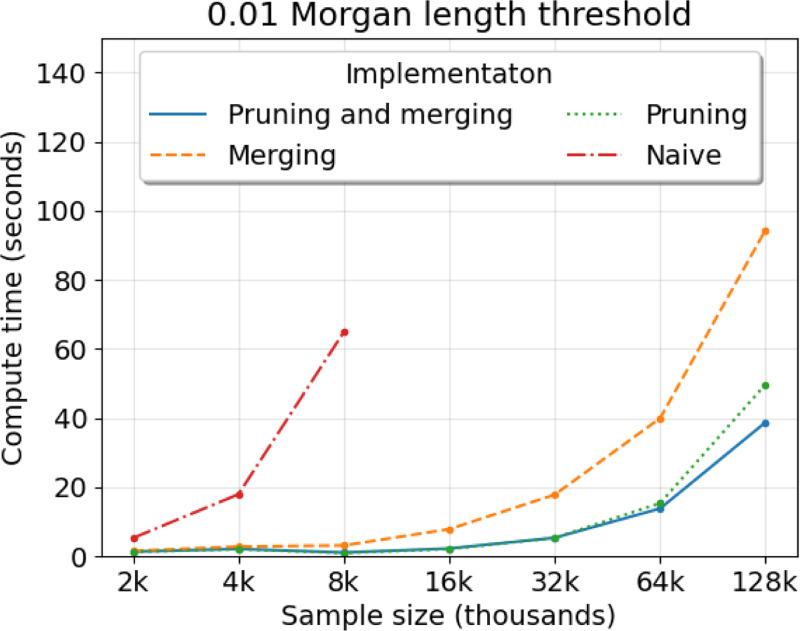
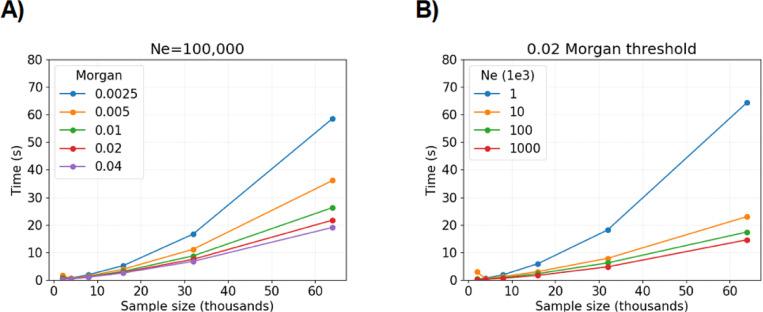
The worst-case runtime complexity to simulate haplotype segments identical by descent (IBD) is quadratic in sample size. We propose two main techniques to reduce the compute time, both of which are motivated by coalescent and recombination processes. We provide mathematical results that explain why our algorithm should outperform a naive implementation with high probability. In our experiments, we observe average compute times to simulate detectable IBD segments around a locus that scale approximately linearly in sample size and take a couple of seconds for sample sizes that are less than ten thousand diploid individuals. In contrast, we find that existing methods to simulate IBD segments take minutes to hours for sample sizes exceeding a few thousand diploid individuals. When using IBD segments to study recent positive selection around a locus, our efficient simulation algorithm makes feasible statistical inferences, e.g., parametric bootstrapping in analyses of large biobanks, that would be otherwise intractable.
Keywords: 60–08; 92D15; 92–04; 92–08; 92–10; coalescent; computational runtime; identity-by-descent.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures


References
-
- Adrion J.R., Cole C.B., Dukler N., Galloway J.G., Gladstein A.L., Gower G., Kyriazis C.C., Ragsdale A.P., Tsambos G., Baumdicker F., Carlson J., Cartwright R.A., Durvasula A., Gronau I., Kim B.Y., McKenzie P., Messer P.W., Noskova E., Ortega-Del Vecchyo D., Racimo F., Struck T.J., Gravel S., Gutenkunst R.N., Lohmueller K.E., Ralph P.L., Schrider D.R., Siepel A., Kelleher J., Kern A.D.: A community-maintained standard library of population genetic models. Elife 9 (2020) - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources