

A dual role of Cohesin in DNA DSB repair
- PMID: 39833168
- PMCID: PMC11747280
- DOI: 10.1038/s41467-025-56086-4
A dual role of Cohesin in DNA DSB repair
Abstract
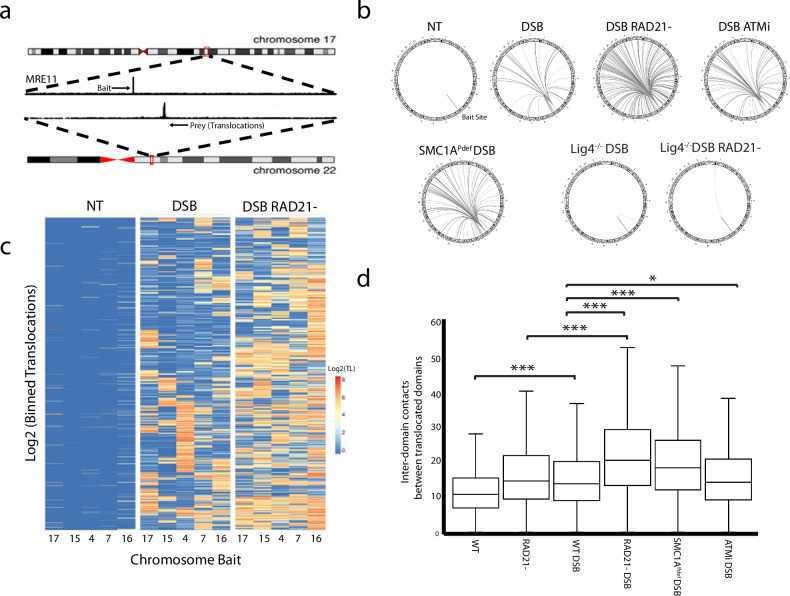
Cells undergo tens of thousands of DNA-damaging events each day. Defects in repairing double-stranded breaks (DSBs) can lead to genomic instability, contributing to cancer, genetic disorders, immunological diseases, and developmental defects. Cohesin, a multi-subunit protein complex, plays a crucial role in both chromosome organization and DNA repair by creating architectural loops through chromatin extrusion. However, the mechanisms by which cohesin regulates these distinct processes are not fully understood. In this study, we identify two separate roles for cohesin in DNA repair within mammalian cells. First, cohesin serves as an intrinsic architectural factor that normally prevents interactions between damaged chromatin. Second, cohesin has an architecture-independent role triggered by ATM phosphorylation of SMC1, which enhances the efficiency of repair. Our findings suggest that these two functions work together to reduce the occurrence of translocations and deletions associated with non-homologous end joining, thereby maintaining genomic stability.
© 2025. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures



References
-
- Curtin, N. J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer12, 801–817 (2012). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
