

Spatially aligned graph transfer learning for characterizing spatial regulatory heterogeneity
- PMID: 39841593
- PMCID: PMC11752617
- DOI: 10.1093/bib/bbaf021
Spatially aligned graph transfer learning for characterizing spatial regulatory heterogeneity
Abstract
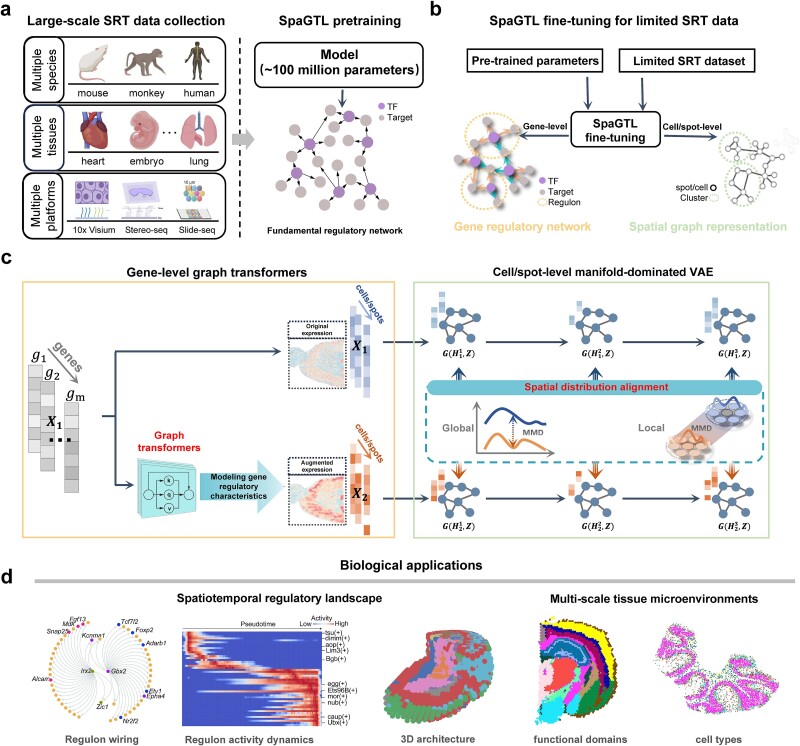
Spatially resolved transcriptomics (SRT) technologies facilitate the exploration of cell fates or states within tissue microenvironments. Despite these advances, the field has not adequately addressed the regulatory heterogeneity influenced by microenvironmental factors. Here, we propose a novel Spatially Aligned Graph Transfer Learning (SpaGTL), pretrained on a large-scale multi-modal SRT data of about 100 million cells/spots to enable inference of context-specific spatial gene regulatory networks across multiple scales in data-limited settings. As a novel cross-dimensional transfer learning architecture, SpaGTL aligns spatial graph representations across gene-level graph transformers and cell/spot-level manifold-dominated variational autoencoder. This alignment facilitates the exploration of microenvironmental variations in cell types and functional domains from a molecular regulatory perspective, all within a self-supervised framework. We verified SpaGTL's precision, robustness, and speed over existing state-of-the-art algorithms and show SpaGTL's potential that facilitates the discovery of novel regulatory programs that exhibit strong associations with tissue functional regions and cell types. Importantly, SpaGTL could be extended to process multi-slice SRT data and map molecular regulatory landscape associated with three-dimensional spatial-temporal changes during development.
Keywords: cross-dimensional transfer learning; graph transformers; spatial regulatory network inference; spatially resolved transcriptomics.
© The Author(s) 2025. Published by Oxford University Press.
Figures




Similar articles
-
Spatially contrastive variational autoencoder for deciphering tissue heterogeneity from spatially resolved transcriptomics.Brief Bioinform. 2024 Jan 22;25(2):bbae016. doi: 10.1093/bib/bbae016. Brief Bioinform. 2024. PMID: 38324623 Free PMC article.
-
Spatially Aware Domain Adaptation Enables Cell Type Deconvolution from Multi-Modal Spatially Resolved Transcriptomics.Small Methods. 2025 May;9(5):e2401163. doi: 10.1002/smtd.202401163. Epub 2024 Dec 2. Small Methods. 2025. PMID: 39623794
-
A multi-view graph contrastive learning framework for deciphering spatially resolved transcriptomics data.Brief Bioinform. 2024 May 23;25(4):bbae255. doi: 10.1093/bib/bbae255. Brief Bioinform. 2024. PMID: 38801701 Free PMC article.
-
Integrating Spatially-Resolved Transcriptomics Data Across Tissues and Individuals: Challenges and Opportunities.Small Methods. 2025 May;9(5):e2401194. doi: 10.1002/smtd.202401194. Epub 2025 Feb 11. Small Methods. 2025. PMID: 39935130 Free PMC article. Review.
-
Computational solutions for spatial transcriptomics.Comput Struct Biotechnol J. 2022 Sep 1;20:4870-4884. doi: 10.1016/j.csbj.2022.08.043. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36147664 Free PMC article. Review.
Cited by
-
Spatially informed graph transformers for spatially resolved transcriptomics.Commun Biol. 2025 Apr 6;8(1):574. doi: 10.1038/s42003-025-08015-w. Commun Biol. 2025. PMID: 40188303 Free PMC article.
References
-
- Chen A, Liao S, Cheng M. et al. . Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 2022;185:e1721. - PubMed
MeSH terms
Grants and funding
- 20224BAB202022/Jiangxi Provincial Natural Science Foundation, China
- GJJ202310/Science and Technology Research Project of Jiangxi Provincial Department of Education, China
- 2023HIAS-Y024/Hangzhou Institute for advanced study, UCAS
- 2024040801020300/Natural Science Foundation of Wuhan
- 62062063/National Natural Science Foundation of China
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
