

Global cellular proteo-lipidomic profiling of diverse lysosomal storage disease mutants using nMOST
- PMID: 39841834
- PMCID: PMC11753374
- DOI: 10.1126/sciadv.adu5787
Global cellular proteo-lipidomic profiling of diverse lysosomal storage disease mutants using nMOST
Abstract
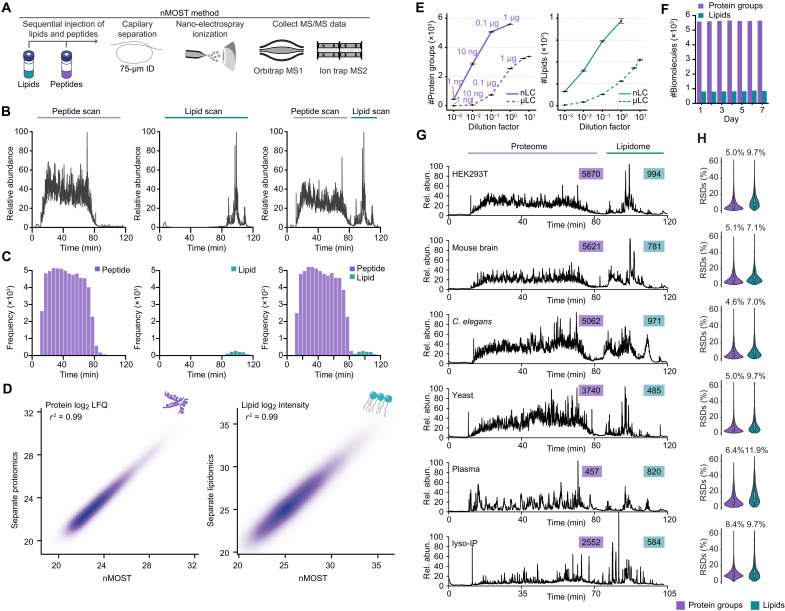
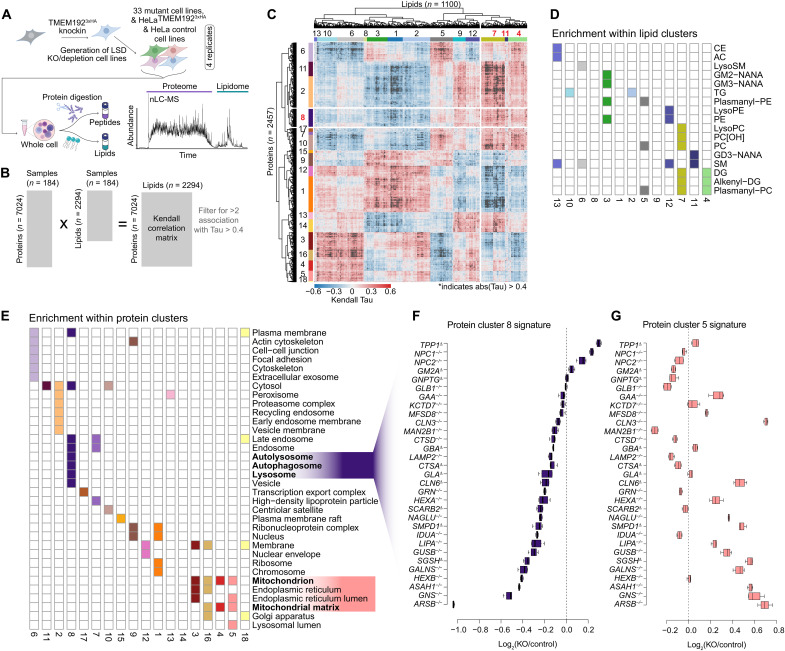
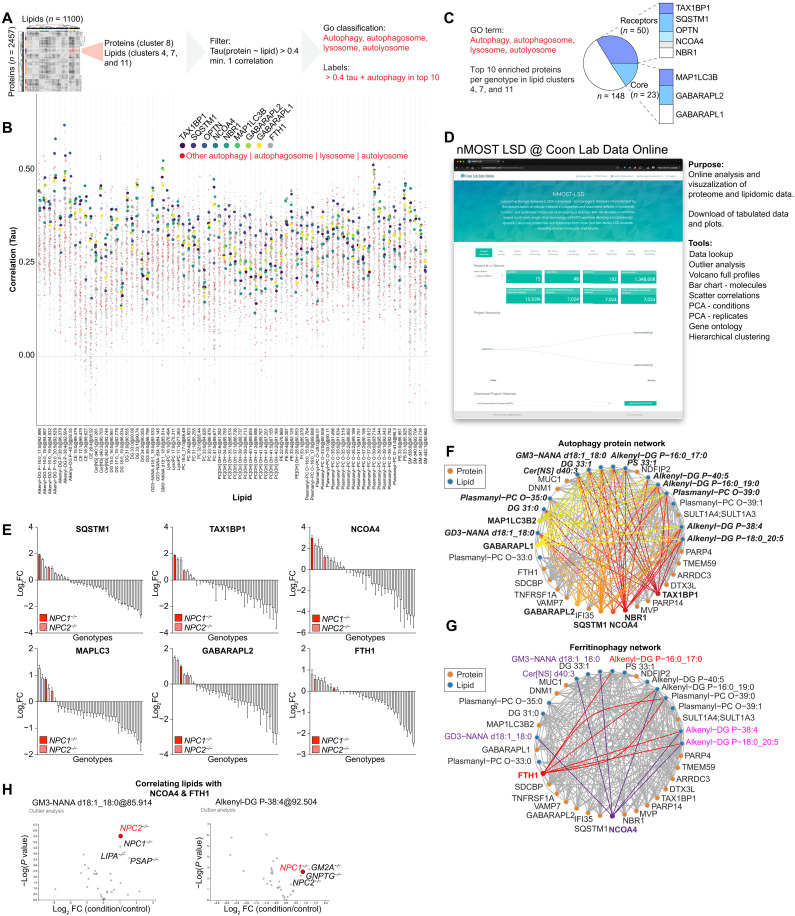
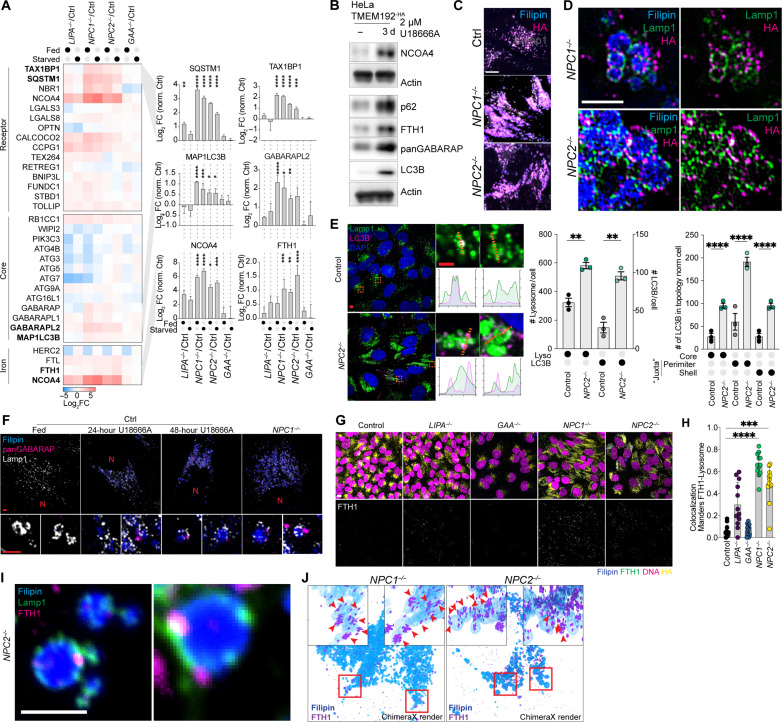
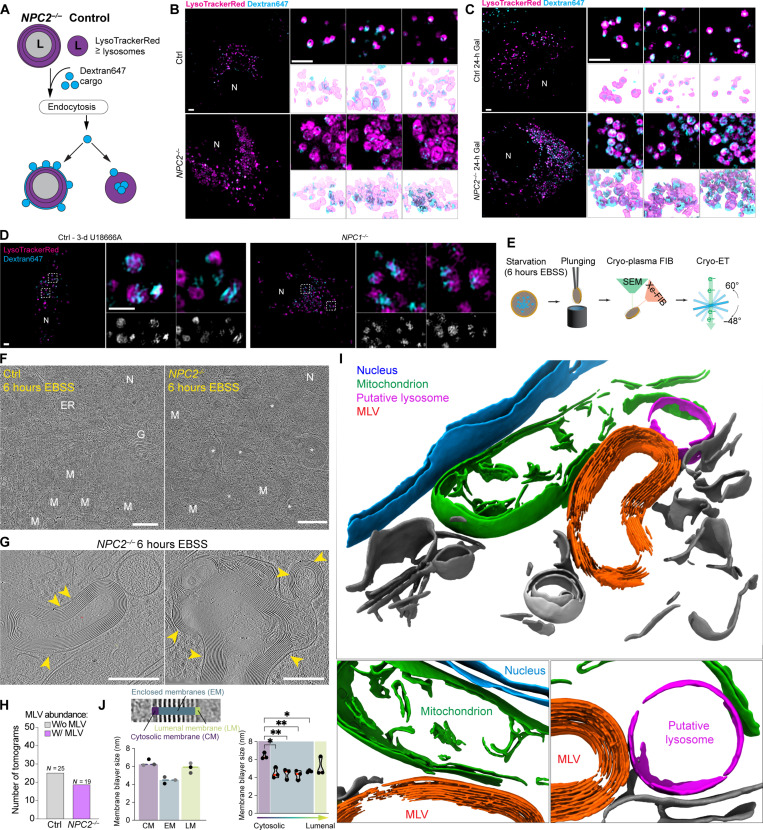
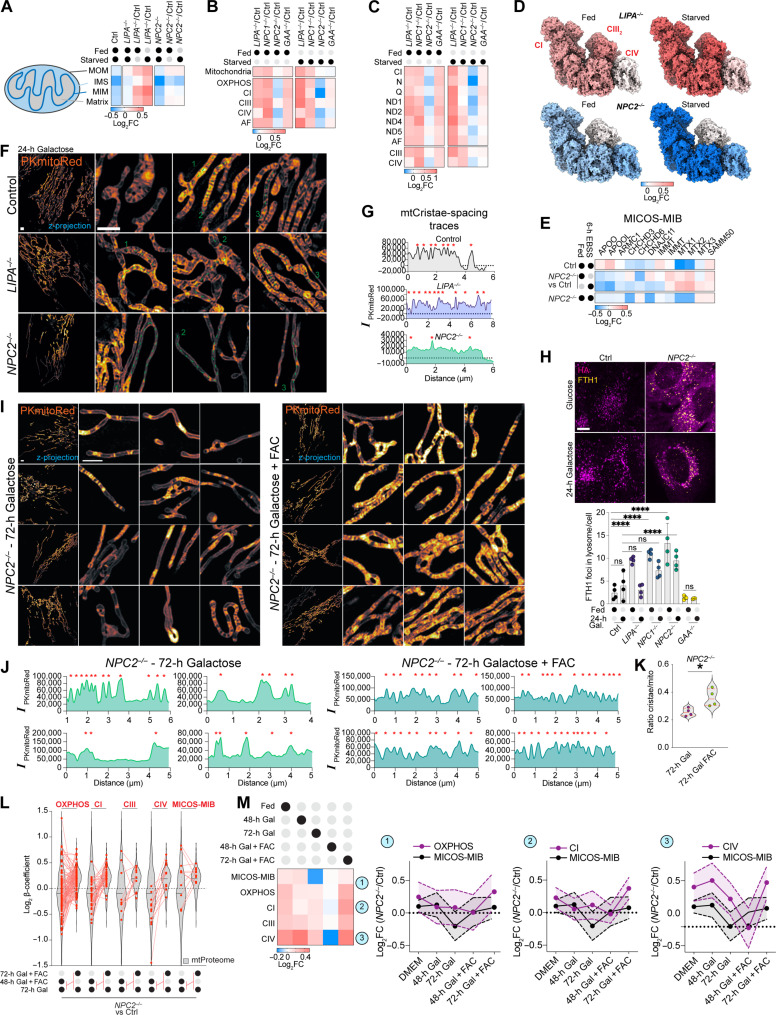
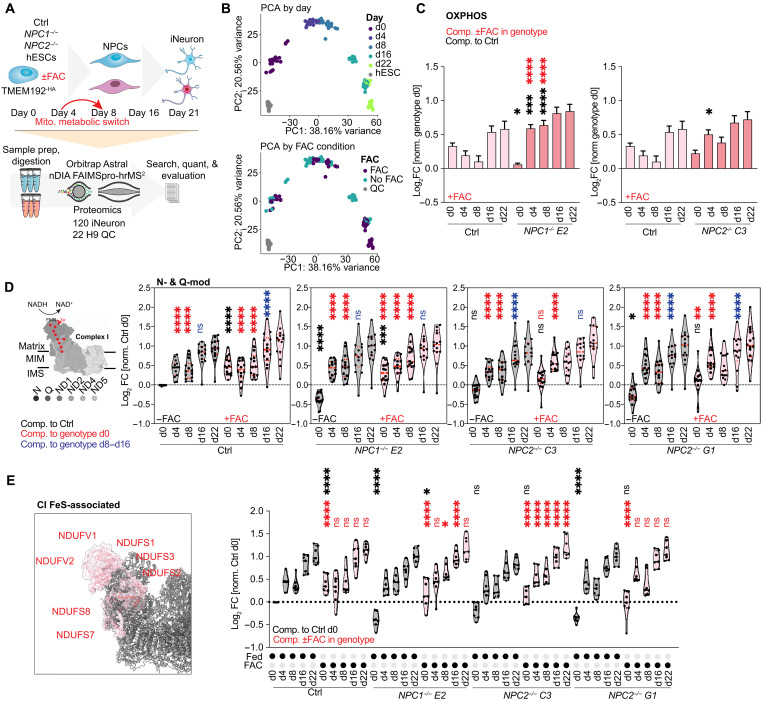
Lysosomal storage diseases (LSDs) comprise ~50 monogenic disorders marked by the buildup of cellular material in lysosomes, yet systematic global molecular phenotyping of proteins and lipids is lacking. We present a nanoflow-based multiomic single-shot technology (nMOST) workflow that quantifies HeLa cell proteomes and lipidomes from over two dozen LSD mutants. Global cross-correlation analysis between lipids and proteins identified autophagy defects, notably the accumulation of ferritinophagy substrates and receptors, especially in NPC1-/- and NPC2-/- mutants, where lysosomes accumulate cholesterol. Autophagic and endocytic cargo delivery failures correlated with elevated lysophosphatidylcholine species and multilamellar structures visualized by cryo-electron tomography. Loss of mitochondrial cristae, MICOS complex components, and OXPHOS components rich in iron-sulfur cluster proteins in NPC2-/- cells was largely alleviated when iron was provided through the transferrin system. This study reveals how lysosomal dysfunction affects mitochondrial homeostasis and underscores nMOST as a valuable discovery tool for identifying molecular phenotypes across LSDs.
Figures
Update of
-
Global cellular proteo-lipidomic profiling of diverse lysosomal storage disease mutants using nMOST.bioRxiv [Preprint]. 2024 Oct 20:2024.03.26.586828. doi: 10.1101/2024.03.26.586828. bioRxiv. 2024. Update in: Sci Adv. 2025 Jan 24;11(4):eadu5787. doi: 10.1126/sciadv.adu5787. PMID: 38585873 Free PMC article. Updated. Preprint.
References
-
- Lawrence R. E., Zoncu R., The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142 (2019). - PubMed
-
- Mutvei A. P., Nagiec M. J., Blenis J., Balancing lysosome abundance in health and disease. Nat. Cell Biol. 25, 1254–1264 (2023). - PubMed
-
- Platt F. M., d'Azzo A., Davidson B. L., Neufeld E. F., Tifft C. J., Lysosomal storage diseases. Nat. Rev. Dis. Primers. 4, 27 (2018). - PubMed
-
- Robak L. A., Jansen I. E., van Rooij J., Uitterlinden A. G., Kraaij R., Jankovic J., International Parkinson’s Disease Genomics Consortium (IPDGC), Heutink P., Shulman J. M., Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain 140, 3191–3203 (2017). - PMC - PubMed
-
- Alcalay R. N., Mallett V., Vanderperre B., Tavassoly O., Dauvilliers Y., Wu R. Y. J., Ruskey J. A., Leblond C. S., Ambalavanan A., Laurent S. B., Spiegelman D., Dionne-Laporte A., Liong C., Levy O. A., Fahn S., Waters C., Kuo S. H., Chung W. K., Ford B., Marder K. S., Kang U. J., Hassin-Baer S., Greenbaum L., Trempe J. F., Wolf P., Oliva P., Zhang X. K., Clark L. N., Langlois M., Dion P. A., Fon E. A., Dupre N., Rouleau G. A., Gan-Or Z., SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson's disease. Mov. Disord. 34, 526–535 (2019). - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
