

In vivo CRISPR-Cas9 genome editing in mice identifies genetic modifiers of somatic CAG repeat instability in Huntington's disease
- PMID: 39843658
- PMCID: PMC11821541
- DOI: 10.1038/s41588-024-02054-5
In vivo CRISPR-Cas9 genome editing in mice identifies genetic modifiers of somatic CAG repeat instability in Huntington's disease
Abstract
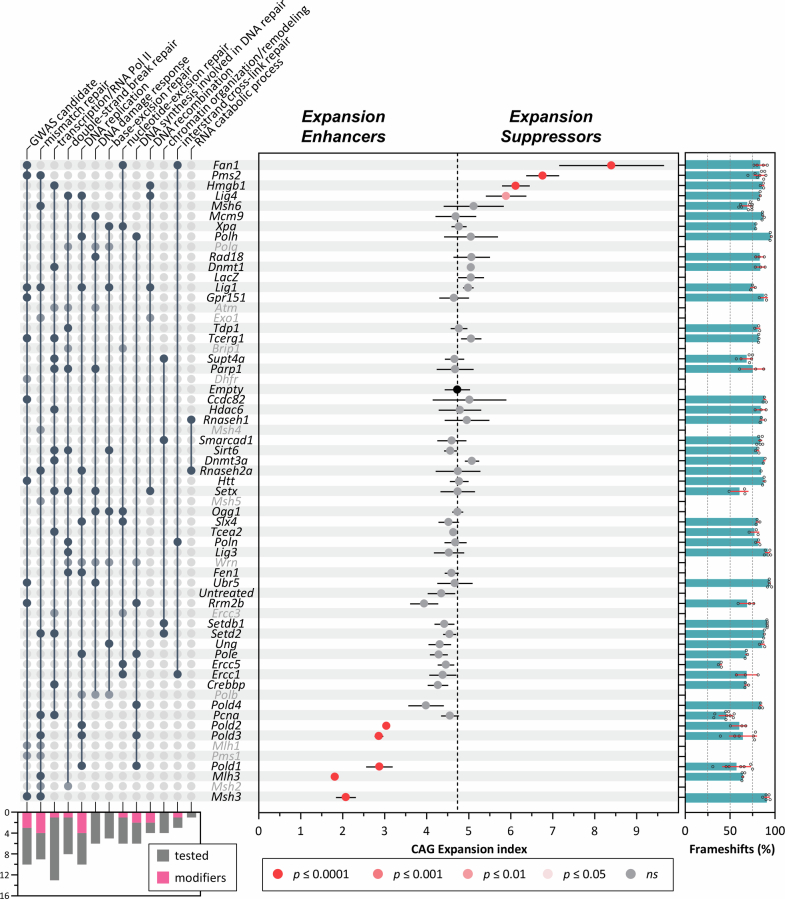
Huntington's disease, one of more than 50 inherited repeat expansion disorders1, is a dominantly inherited neurodegenerative disease caused by a CAG expansion in HTT2. Inherited CAG repeat length is the primary determinant of age of onset, with human genetic studies underscoring that the disease is driven by the CAG length-dependent propensity of the repeat to further expand in the brain3-9. Routes to slowing somatic CAG expansion, therefore, hold promise for disease-modifying therapies. Several DNA repair genes, notably in the mismatch repair pathway, modify somatic expansion in Huntington's disease mouse models10. To identify novel modifiers of somatic expansion, we used CRISPR-Cas9 editing in Huntington's disease knock-in mice to enable in vivo screening of expansion-modifier candidates at scale. This included testing of Huntington's disease onset modifier genes emerging from human genome-wide association studies as well as interactions between modifier genes, providing insight into pathways underlying CAG expansion and potential therapeutic targets.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: R.M.P. and V.C.W. received research support from Pfizer for this study. J.F.G. and V.C.W. were founding scientific advisory board members with a financial interest in Triplet Therapeutics. Their financial interests were reviewed and are managed by MGH and Mass General Brigham (MGB) in accordance with their conflict-of-interest policies. V.C.W. is a scientific advisory board member of LoQus23 Therapeutics and has provided paid consulting services to Acadia Pharmaceuticals, Alnylam, Biogen, Passage Bio and Rgenta Therapeutics. J.F.G. consults for Transine Therapeutics (dba Harness Therapeutics) and has previously provided paid consulting services to Wave Therapeutics USA, Biogen and Pfizer. The other authors declare no competing interests.
Figures















Update of
-
Identification of genetic modifiers of Huntington's disease somatic CAG repeat instability by in vivo CRISPR-Cas9 genome editing.bioRxiv [Preprint]. 2024 Jun 9:2024.06.08.597823. doi: 10.1101/2024.06.08.597823. bioRxiv. 2024. Update in: Nat Genet. 2025 Feb;57(2):314-322. doi: 10.1038/s41588-024-02054-5. PMID: 38895438 Free PMC article. Updated. Preprint.
References
-
- The Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell72, 971–983 (1993). - PubMed
MeSH terms
Substances
Grants and funding
- NS111066/U.S. Department of Health & Human Services | NIH | Office of Extramural Research, National Institutes of Health (OER)
- NS049206/U.S. Department of Health & Human Services | NIH | Office of Extramural Research, National Institutes of Health (OER)
- NS091161/U.S. Department of Health & Human Services | NIH | Office of Extramural Research, National Institutes of Health (OER)
- R01 NS091161/NS/NINDS NIH HHS/United States
- R01 NS126420/NS/NINDS NIH HHS/United States
- R01 NS049206/NS/NINDS NIH HHS/United States
- NS126420/U.S. Department of Health & Human Services | NIH | Office of Extramural Research, National Institutes of Health (OER)
- P30 CA006516/CA/NCI NIH HHS/United States
- R21 NS111066/NS/NINDS NIH HHS/United States
- NS049206/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
