

Multiscale footprints reveal the organization of cis-regulatory elements
- PMID: 39843737
- PMCID: PMC11839466
- DOI: 10.1038/s41586-024-08443-4
Multiscale footprints reveal the organization of cis-regulatory elements
Abstract
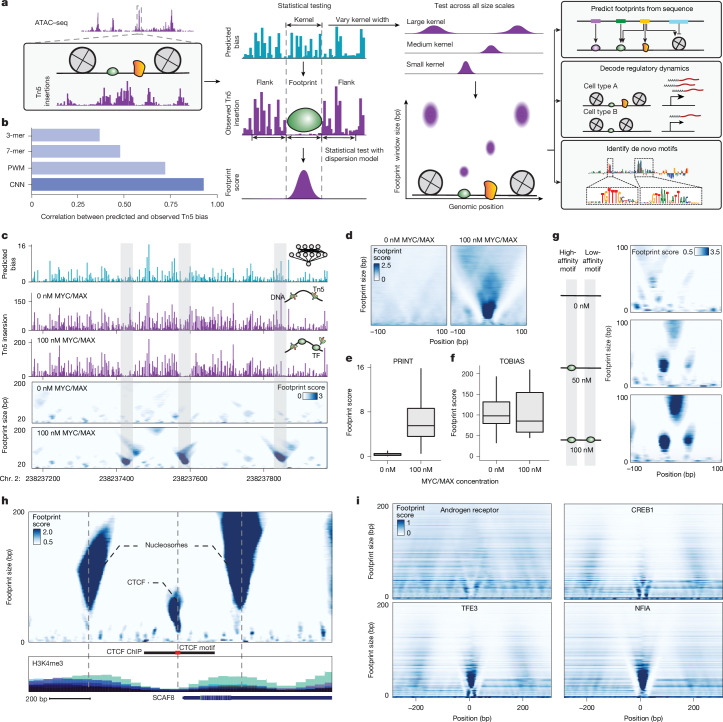
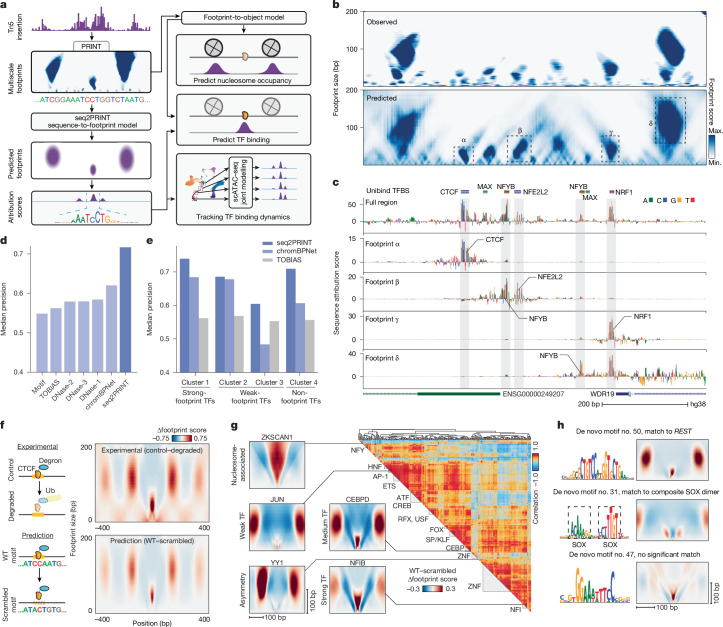
Cis-regulatory elements (CREs) control gene expression and are dynamic in their structure and function, reflecting changes in the composition of diverse effector proteins over time1. However, methods for measuring the organization of effector proteins at CREs across the genome are limited, hampering efforts to connect CRE structure to their function in cell fate and disease. Here we developed PRINT, a computational method that identifies footprints of DNA-protein interactions from bulk and single-cell chromatin accessibility data across multiple scales of protein size. Using these multiscale footprints, we created the seq2PRINT framework, which uses deep learning to allow precise inference of transcription factor and nucleosome binding and interprets regulatory logic at CREs. Applying seq2PRINT to single-cell chromatin accessibility data from human bone marrow, we observe sequential establishment and widening of CREs centred on pioneer factors across haematopoiesis. We further discover age-associated alterations in the structure of CREs in murine haematopoietic stem cells, including widespread reduction of nucleosome footprints and gain of de novo identified Ets composite motifs. Collectively, we establish a method for obtaining rich insights into DNA-binding protein dynamics from chromatin accessibility data, and reveal the architecture of regulatory elements across differentiation and ageing.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: J.D.B. holds patents related to ATAC–seq, is a consultant for the Treehouse Family Foundation, and is a SAB member of Camp4 and seqWell. J.D.B. and S.M. hold a patent based on SHARE–seq. M.A.H. holds patents related to CRISPR-mediated interference and activation, and is a consultant for Akuous, Inc., DEM Biopharma and Gordian Biotechnology. A.J.W. is a scientific advisor for Frequency Therapeutics and Kate Therapeutics, is also a cofounder and scientific advisory board member and holds private equity in Elevian, Inc., a company that aims to develop medicines to restore regenerative capacity. Elevian also provides sponsored research to the Wagers laboratory. The other authors declare no competing interests.
Figures












References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
