

Mapping cells through time and space with moscot
- PMID: 39843746
- PMCID: PMC11864987
- DOI: 10.1038/s41586-024-08453-2
Mapping cells through time and space with moscot
Abstract
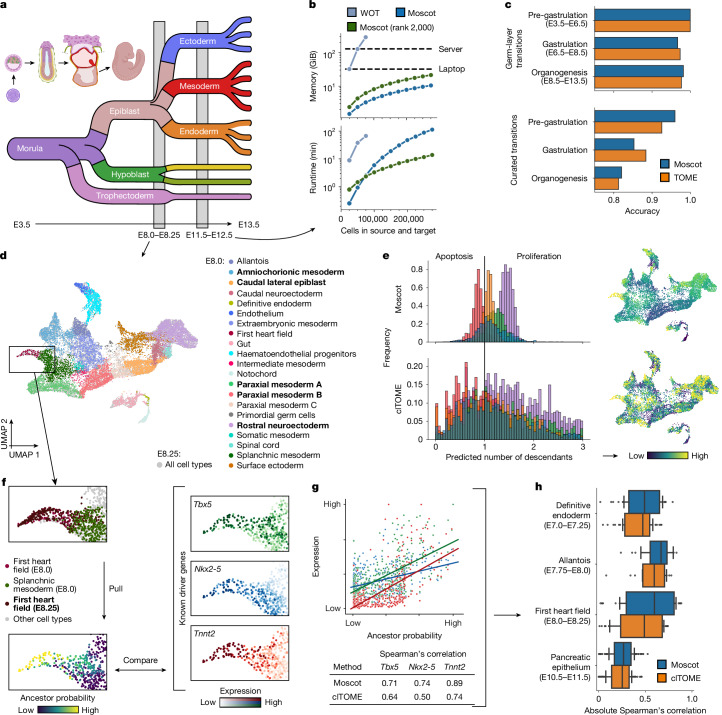
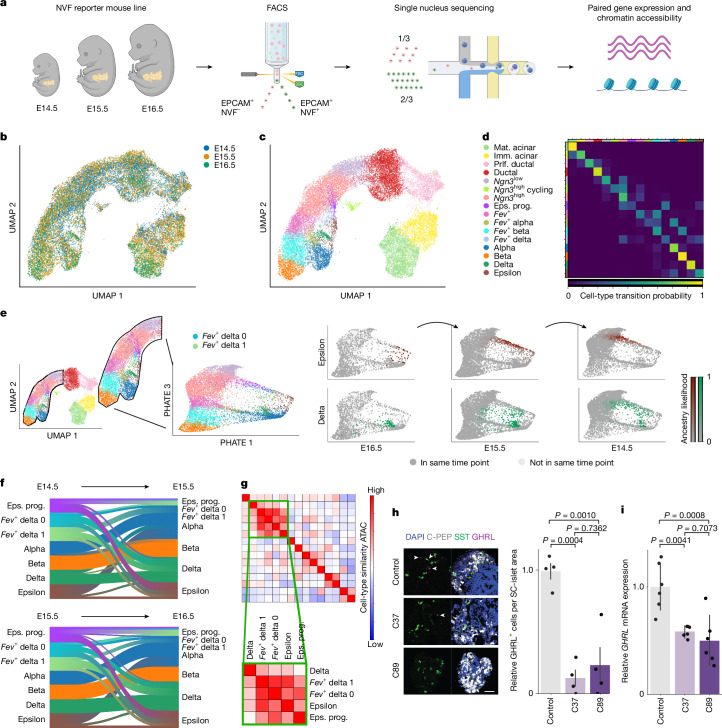
Single-cell genomic technologies enable the multimodal profiling of millions of cells across temporal and spatial dimensions. However, experimental limitations hinder the comprehensive measurement of cells under native temporal dynamics and in their native spatial tissue niche. Optimal transport has emerged as a powerful tool to address these constraints and has facilitated the recovery of the original cellular context1-4. Yet, most optimal transport applications are unable to incorporate multimodal information or scale to single-cell atlases. Here we introduce multi-omics single-cell optimal transport (moscot), a scalable framework for optimal transport in single-cell genomics that supports multimodality across all applications. We demonstrate the capability of moscot to efficiently reconstruct developmental trajectories of 1.7 million cells from mouse embryos across 20 time points. To illustrate the capability of moscot in space, we enrich spatial transcriptomic datasets by mapping multimodal information from single-cell profiles in a mouse liver sample and align multiple coronal sections of the mouse brain. We present moscot.spatiotemporal, an approach that leverages gene-expression data across both spatial and temporal dimensions to uncover the spatiotemporal dynamics of mouse embryogenesis. We also resolve endocrine-lineage relationships of delta and epsilon cells in a previously unpublished mouse, time-resolved pancreas development dataset using paired measurements of gene expression and chromatin accessibility. Our findings are confirmed through experimental validation of NEUROD2 as a regulator of epsilon progenitor cells in a model of human induced pluripotent stem cell islet cell differentiation. Moscot is available as open-source software, accompanied by extensive documentation.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: F.J.T. consults for Immunai, Singularity Bio, CytoReason, Cellarity and Omniscope, and has ownership interest in Dermagnostix and Cellarity. The remaining authors declare no competing interests.
Figures













References
-
- Peyré, G. & Cuturi, M. Computational Optimal Transport: With Applications to Data Science (Now Publishers, 2019) 10.1561/9781680835519.
-
- Cuturi, M. Sinkhorn distances: lightspeed computation of optimal transport. In Advances in Neural information Processing Systems 26 (eds Burges, C. J. et al.) 1– 9 (Curran Associates Inc., 2013).
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
