

Tousled-like kinase loss confers PARP inhibitor resistance in BRCA1-mutated cancers by impeding non-homologous end joining repair
- PMID: 39844055
- PMCID: PMC11753094
- DOI: 10.1186/s10020-025-01066-z
Tousled-like kinase loss confers PARP inhibitor resistance in BRCA1-mutated cancers by impeding non-homologous end joining repair
Abstract
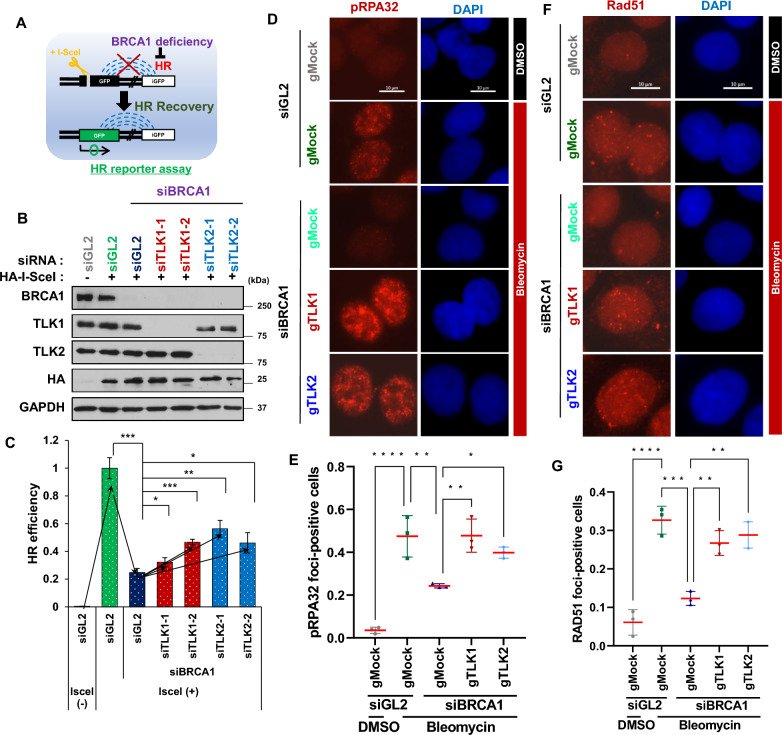
Background: Double-strand breaks (DSBs) are primarily repaired through non-homologous end joining (NHEJ) and homologous recombination (HR). Given that DSBs are highly cytotoxic, PARP inhibitors (PARPi), a prominent class of anticancer drugs, are designed to target tumors with HR deficiency (HRD), such as those harboring BRCA mutations. However, many tumor cells acquire resistance to PARPi, often by restoring HR in HRD cells through the inactivation of NHEJ. Therefore, identifying novel regulators of NHEJ could provide valuable insights into the mechanisms underlying PARPi resistance.
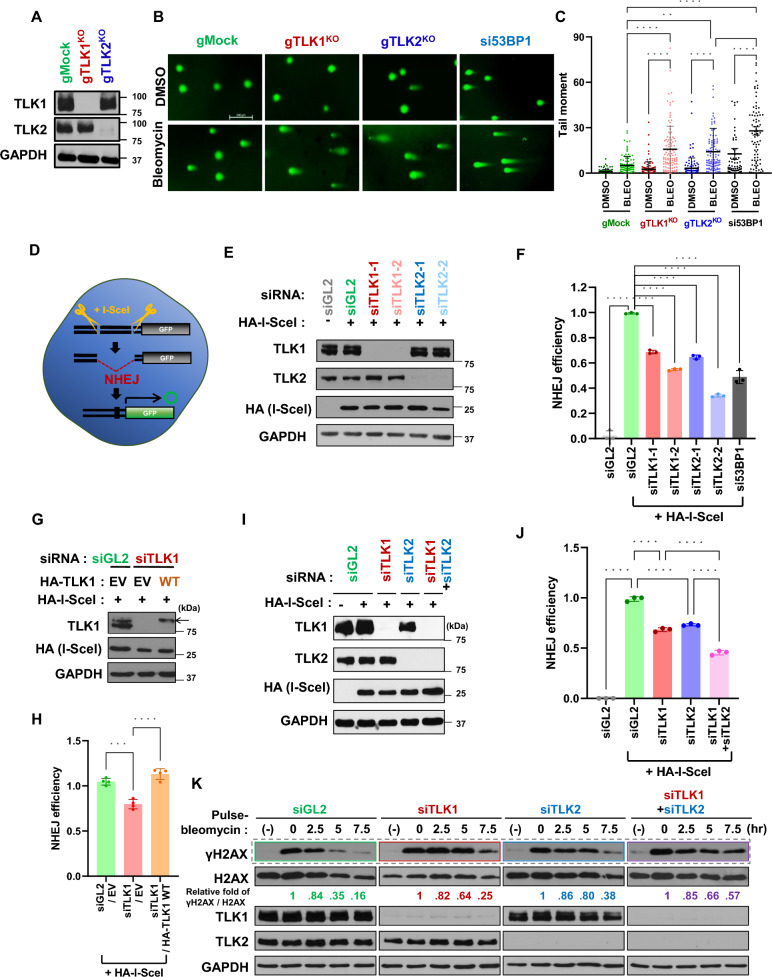
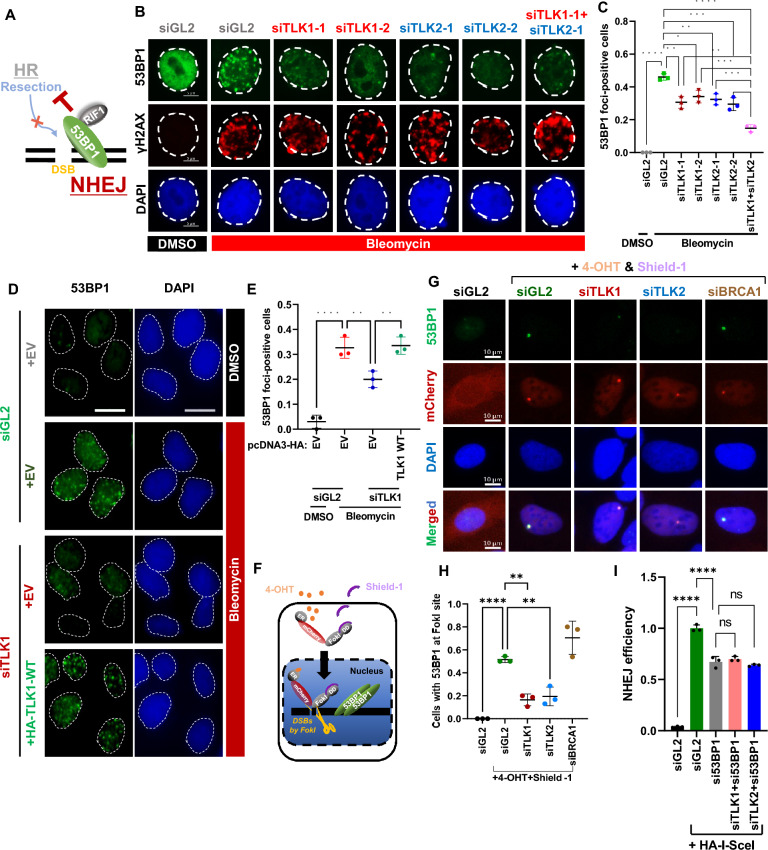
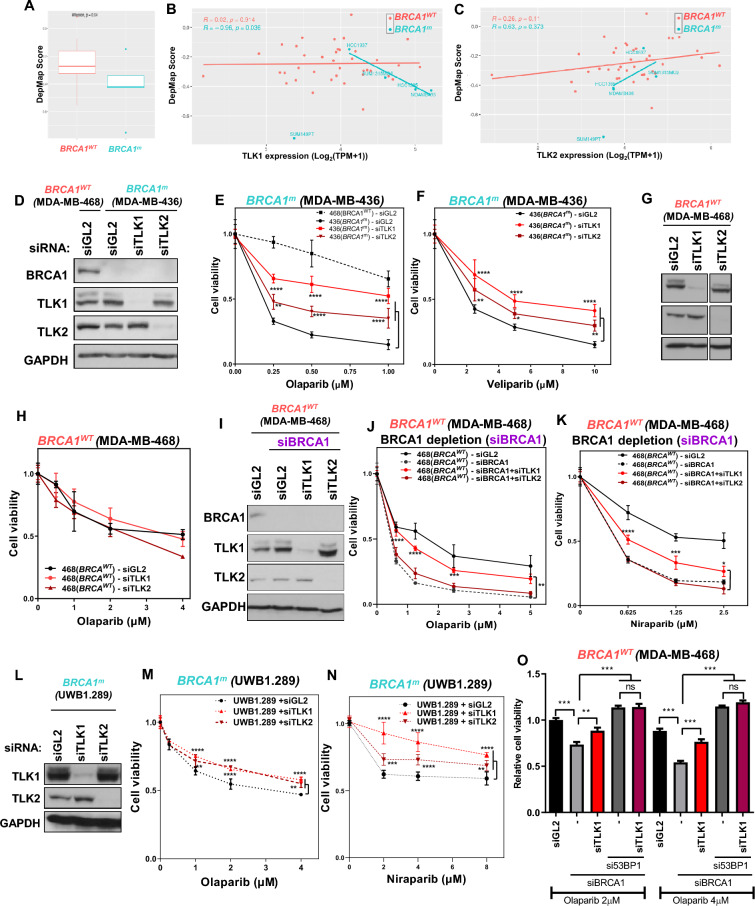
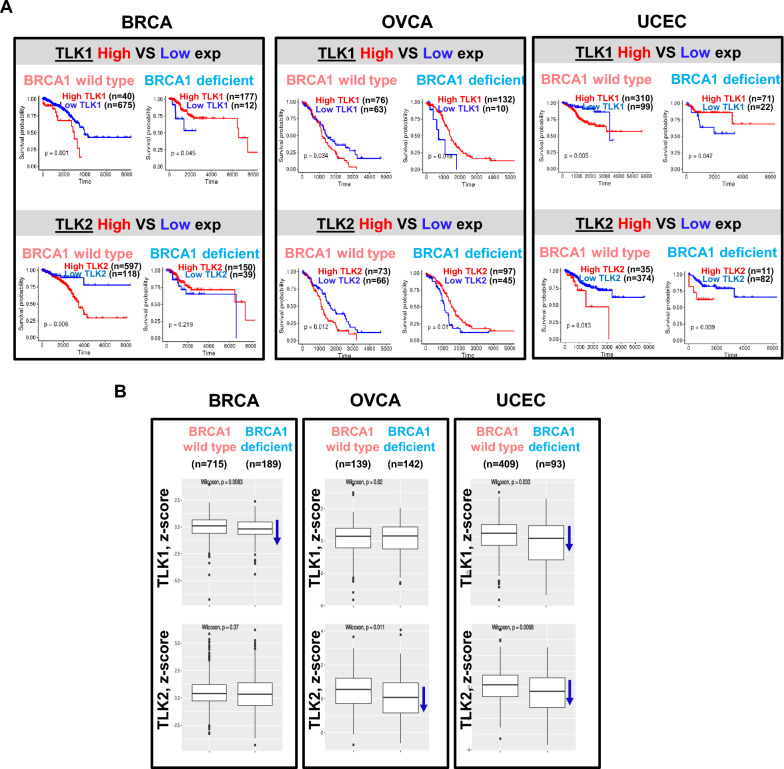
Methods: Cellular DSBs were assessed using neutral comet assays and phospho-H2AX immunoblotting. Fluorescence-based reporter assays quantified repair via NHEJ or HR. The recruitment of proteins that promote NHEJ and HR to DSBs was analyzed using immunostaining, live-cell imaging following laser-induced microirradiation, and FokI-inducible single DSB generation. Loss-of-function experiments were performed in multiple human cancer cell lines using siRNA-mediated knockdown or CRISPR-Cas9 gene knockout. Cell viability assays were conducted to evaluate resistance to PARP inhibitors. Additionally, bioinformatic analyses of public databases were performed to investigate the association between TLK expression and BRCA1 status.
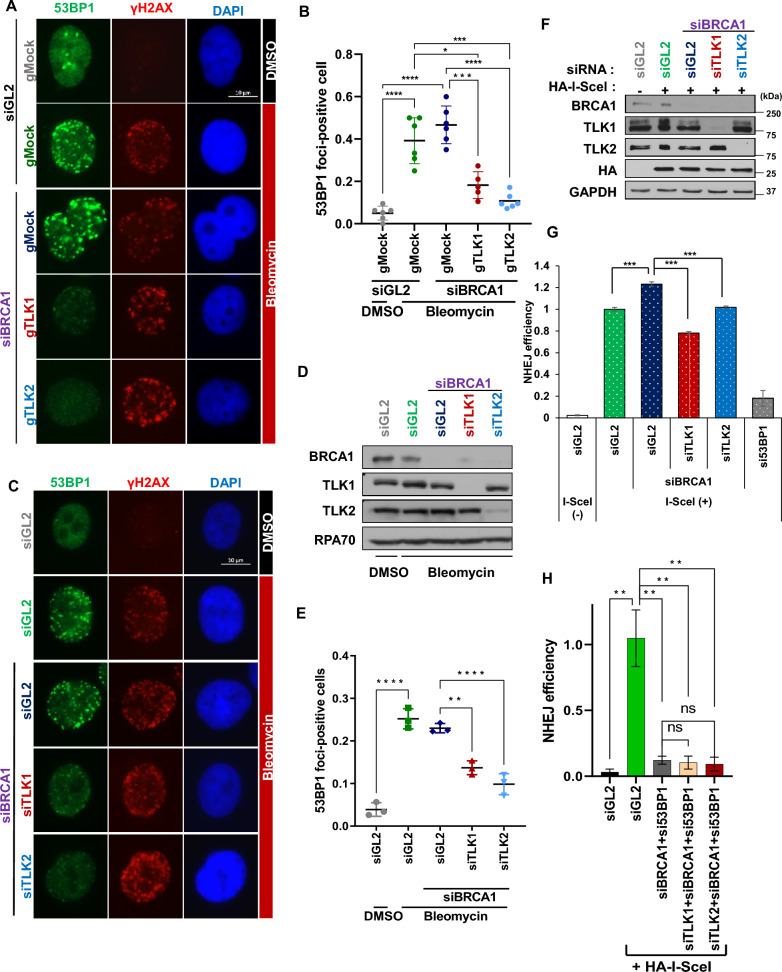
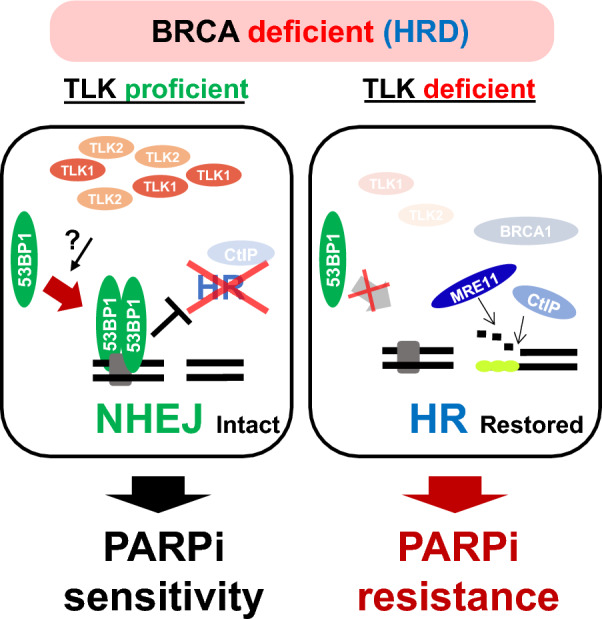
Results: We demonstrate that human tousled-like kinase (TLK) orthologs are essential for NHEJ-mediated repair of DSBs and for PARPi sensitivity in cells with BRCA1 mutation. TLK1 and TLK2 exhibit redundant roles in promoting NHEJ, and their deficiency results in a significant accumulation of DSBs. TLKs are required for the proper localization of 53BP1, a key factor in promoting the NHEJ pathway. Consequently, TLK deficiency induces PARPi resistance in triple-negative breast cancer (TNBC) and ovarian cancer (OVCA) cell lines with BRCA1 deficiency, as TLK deficiency in BRCA1-depleted cells, impairs 53BP1 recruitment to DSBs and reduces NHEJ efficiency, while restoring HR.
Conclusions: We have identified TLK proteins as novel regulators of NHEJ repair and PARPi sensitivity in BRCA1-depleted cells, suggesting that TLK repression may represent a previously unrecognized mechanism by which BRCA1 mutant cancers acquire PARPi resistance.
Keywords: BRCA1; Cancer Therapy; Cancer treatment; DNA damage repair; Double-stranded breaks (DSBs); Drug resistance; Homologous recombination (HR); Non-homologous end joining (NHEJ); PARP inhibitor; Tousled-like kinase.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent of publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures
References
-
- Belal H, Ying Ng EF, Meitinger F. 53BP1-mediated activation of the tumor suppressor p53. Curr Opin Cell Biol. 2024;91: 102424. - PubMed
-
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
