

Deciphering hub genes and immune landscapes related to neutrophil extracellular traps in rheumatoid arthritis: insights from integrated bioinformatics analyses and experiments
- PMID: 39845946
- PMCID: PMC11750673
- DOI: 10.3389/fimmu.2024.1521634
Deciphering hub genes and immune landscapes related to neutrophil extracellular traps in rheumatoid arthritis: insights from integrated bioinformatics analyses and experiments
Abstract
Background: Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by synovial inflammation and progressive joint destruction. Neutrophil extracellular traps (NETs), a microreticular structure formed after neutrophil death, have recently been implicated in RA pathogenesis and pathological mechanisms. However, the underlying molecular mechanisms and key genes involved in NET formation in RA remain largely unknown.
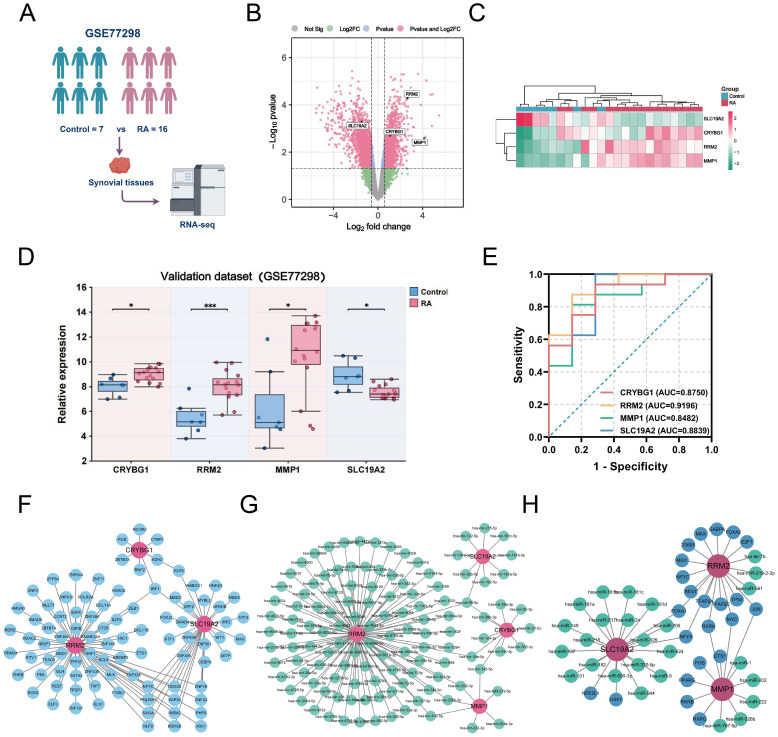
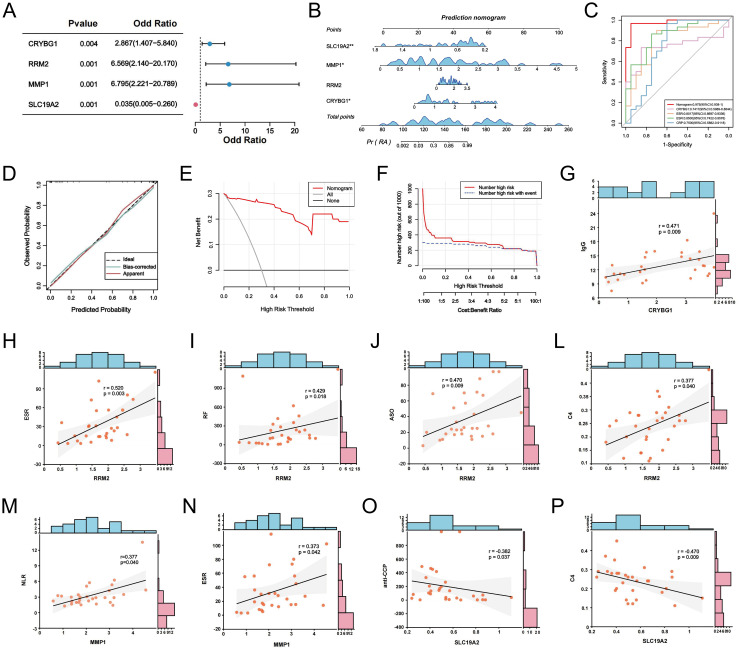
Methods: We obtained single-cell RNA sequencing data of synovial tissues from the Gene Expression Omnibus (GEO) database and performed cellular annotation and intercellular communication analyses. Subsequently, three microarray datasets were collected for a training cohort and correlated with a bulk RNA-seq dataset associated with NETs. Differentially expressed genes were identified, and weighted gene correlation network analysis was used to characterize gene association. Using three machine learning techniques, we identified the most important hub genes to develop and evaluate a nomogram diagnostic model. CIBERSORT was used to elucidate the relationship between hub genes and immune cells. An external validation dataset was used to verify pivotal gene expression and to construct co-regulatory networks using the NetworkAnalyst platform. We further investigated hub gene expression using immunohistochemistry (IHC) in an adjuvant-induced arthritis rat model and real-time quantitative polymerase chain reaction (RT-qPCR) in a clinical cohort.
Results: Seven cellular subpopulations were identified through downscaling and clustering, with neutrophils likely the most crucial cell clusters in RA. Intercellular communication analysis highlighted the network between neutrophils and fibroblasts. In this context, 4 key hub genes (CRYBG1, RMM2, MMP1, and SLC19A2) associated with NETs were identified. A nomogram model with a diagnostic value was developed and evaluated. Immune cell infiltration analysis indicated associations between the hub genes and the immune landscape in NETs and RA. IHC and RT-qPCR findings showed high expression of CRYBG1, RMM2, and MMP1 in synovial and neutrophilic cells, with lower expression of SLC19A2. Correlation analysis further emphasized close associations between hub genes and laboratory markers in patients with RA.
Conclusion: This study first elucidated neutrophil heterogeneity in the RA synovial microenvironment and mechanisms of communication with fibroblasts. CRYBG1, RMM2, MMP1, and SLC19A2 were identified and validated as potential NET-associated biomarkers, offering insights for diagnostic tools and immunotherapeutic strategies in RA.
Keywords: inflammation; neutrophil extracellular traps; neutrophils; rheumatoid arthritis; synovial microenvironment; systems bioinformatics.
Copyright © 2025 Li, Liu, Sun, Hu, Zhou, Cong and Chen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures









Similar articles
-
Exploration and Identification of Potential Biomarkers and Immune Cell Infiltration Analysis in Synovial Tissue of Rheumatoid Arthritis.Int J Rheum Dis. 2025 Feb;28(2):e70137. doi: 10.1111/1756-185X.70137. Int J Rheum Dis. 2025. PMID: 39953769
-
Machine learning and weighted gene co-expression network analysis identify a three-gene signature to diagnose rheumatoid arthritis.Front Immunol. 2024 Apr 22;15:1387311. doi: 10.3389/fimmu.2024.1387311. eCollection 2024. Front Immunol. 2024. PMID: 38711508 Free PMC article.
-
Identification of Disease-Specific Hub Biomarkers and Immune Infiltration in Osteoarthritis and Rheumatoid Arthritis Synovial Tissues by Bioinformatics Analysis.Dis Markers. 2021 May 17;2021:9911184. doi: 10.1155/2021/9911184. eCollection 2021. Dis Markers. 2021. PMID: 34113405 Free PMC article.
-
Neutrophils in the Pathogenesis of Rheumatoid Arthritis and Systemic Lupus Erythematosus: Same Foe Different M.O.Front Immunol. 2021 Mar 4;12:649693. doi: 10.3389/fimmu.2021.649693. eCollection 2021. Front Immunol. 2021. PMID: 33746988 Free PMC article. Review.
-
The emerging role of neutrophil extracellular traps in the progression of rheumatoid arthritis.Front Immunol. 2024 Aug 16;15:1438272. doi: 10.3389/fimmu.2024.1438272. eCollection 2024. Front Immunol. 2024. PMID: 39221253 Free PMC article. Review.
Cited by
-
Neutrophils: a Central Point of Interaction Between Immune Cells and Nonimmune Cells in Rheumatoid Arthritis.Clin Rev Allergy Immunol. 2025 Mar 28;68(1):34. doi: 10.1007/s12016-025-09044-3. Clin Rev Allergy Immunol. 2025. PMID: 40148714 Review.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Medical