

DNA Damage-Induced Ferroptosis: A Boolean Model Regulating p53 and Non-Coding RNAs in Drug Resistance
- PMID: 39846637
- PMCID: PMC11755436
- DOI: 10.3390/proteomes13010006
DNA Damage-Induced Ferroptosis: A Boolean Model Regulating p53 and Non-Coding RNAs in Drug Resistance
Abstract
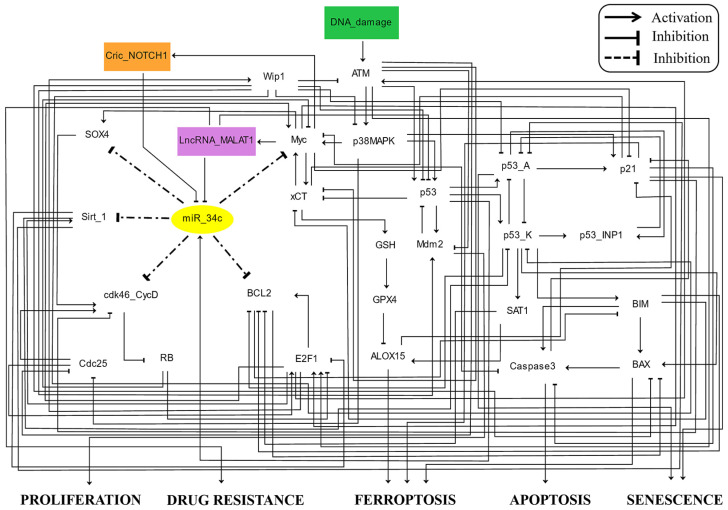
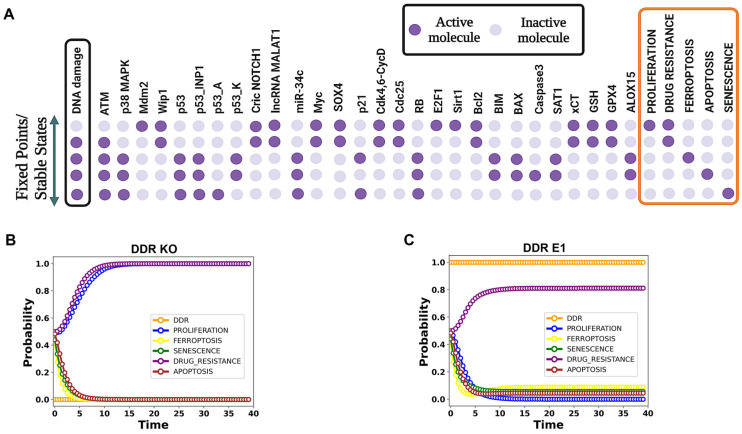
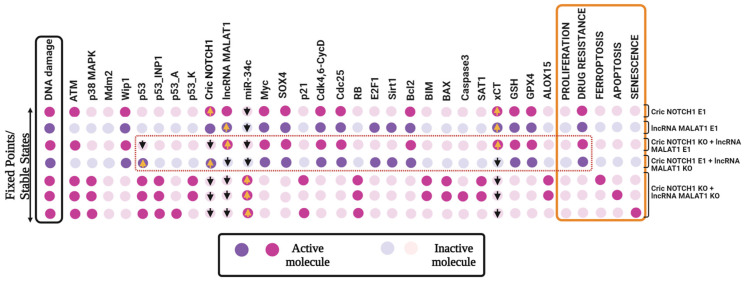
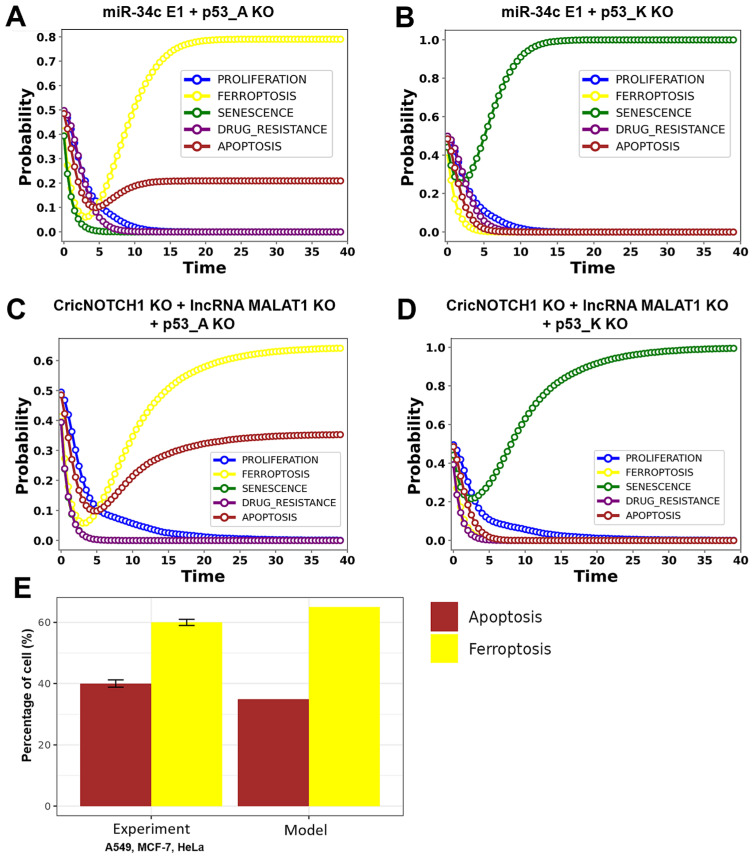
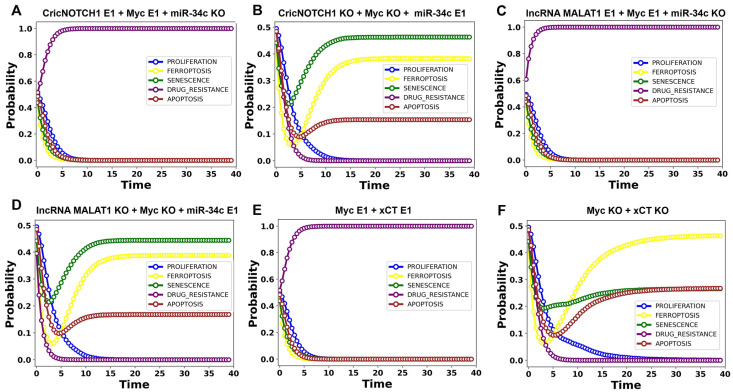
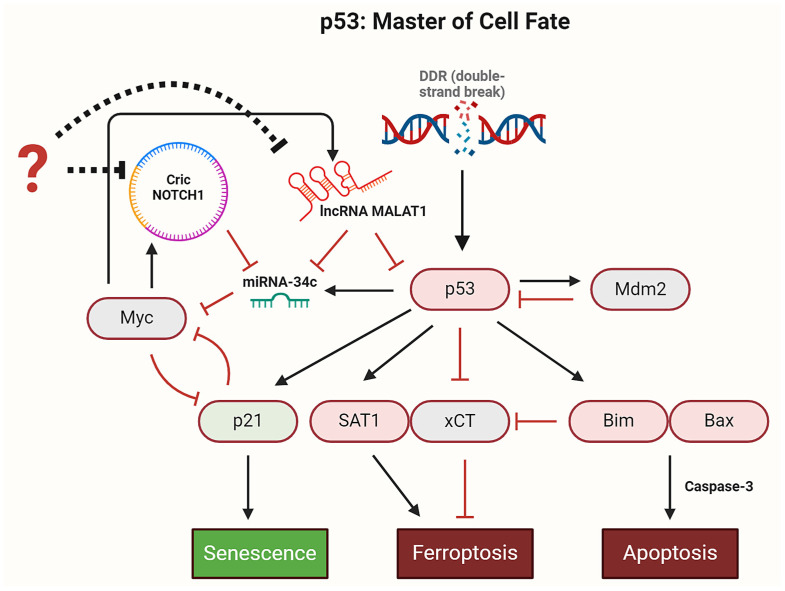
The tumor suppressor p53, in its wild-type form, plays a central role in cellular homeostasis by regulating senescence, apoptosis, and autophagy within the DNA damage response (DDR). Recent findings suggest that wild-type p53 also governs ferroptosis, an iron-dependent cell death process driven by lipid peroxidation. Post-translational modifications of p53 generate proteoforms that significantly enhance its functional diversity in regulating these mechanisms. A key target in this process is the cystine/glutamate transporter (xCT), which is essential for redox balance and ferroptosis resistance. Additionally, p53-induced miR-34c-5p suppresses cancer cell proliferation and drug resistance by modulating Myc, an oncogene further influenced by non-coding RNAs like circular RNA NOTCH1 (CricNOTCH1) and long non-coding RNA MALAT1. However, the exact role of these molecules in ferroptosis remains unclear. To address this, we introduce the first dynamic Boolean model that delineates the influence of these ncRNAs and p53 on ferroptosis, apoptosis, and senescence within the DDR context. Validated through gain- and loss-of-function perturbations, our model closely aligns with experimental observations in cancers such as oral squamous cell carcinoma, nasopharyngeal carcinoma, and osteosarcoma. The model identifies crucial positive feedback loops (CricNOTCH1/miR-34c/Myc, MALAT1/miR-34c/Myc, and Myc/xCT) and highlights the therapeutic potential of using p53 proteoforms and ncRNAs to combat drug resistance and induce cancer cell death.
Keywords: CricRNA NOTCH1; apoptosis; ferroptosis; lncRNA MALAT1; miR-34c-5p; p53.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
