

Clinical manifestations in Egyptian Pompe disease patients: Molecular variability and enzyme replacement therapy (ERT) outcomes
- PMID: 39849595
- PMCID: PMC11756172
- DOI: 10.1186/s13052-025-01837-8
Clinical manifestations in Egyptian Pompe disease patients: Molecular variability and enzyme replacement therapy (ERT) outcomes
Abstract
Background: Pompe disease is a rare genetic disorder caused by a deficiency of the enzyme acid alpha-glucosidase. This condition leads to muscle weakness, respiratory problems, and heart abnormalities in affected individuals.
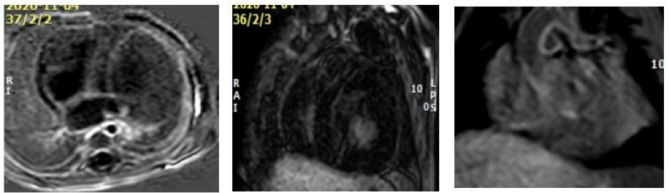
Methods: The aim of the study is to share our experience through cross sectional study of patients with infantile-onset Pompe disease (IOPD) with different genetic variations, resulting in diverse clinical presentations. We evaluated their phenotype, genotype, radiological and laboratory findings including their cross-reactive immunologic material (CRIM) status. Infantile Pompe disease was diagnosed by measurement of the activity of the enzyme alpha-glucosidase. The diagnosis was confirmed by molecular genetic testing using PCR amplification and sequencing of the acid alpha-glucosidase (GAA) gene. Routine two-D echocardiography, and multi-parametric ECG-gated cardiac magnetic resonance imaging (CMR) were done to patients six months after starting enzyme replacement therapy (ERT).
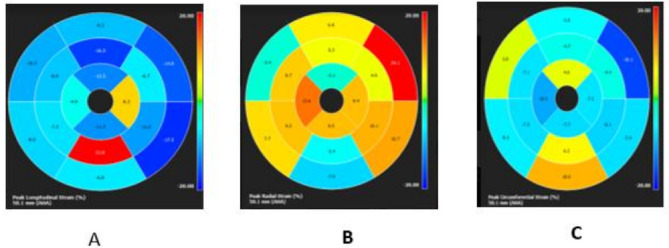
Results: The results of our study revealed different genetic mutations among our patients, different CRIM status and also CMR abnormalities. CMR imaging revealed abnormalities in all cases that underwent the procedure, including myocardial and vascular changes, with feature tracking indicating issues across all parameters and LGE suggesting fibrosis. The patient with a positive immune response had the most severe cardiac abnormalities, despite improvements in muscle weakness and motor skills from ERT. This underscores that delayed diagnosis and ERT can lead to irreversible heart damage from autophagy buildup.
Conclusion: Pompe disease has various clinical presentations and results in significant CMR findings, which can be attributed to different genetic mutations. Early initiation of enzyme replacement therapy in infantile-onset Pompe disease is important to maximize its benefits.
Keywords: Cardiac magnetic resonance imaging (CMR); ERT; Genotypes; Pompe disease.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: All experimental protocols were approved from ethical committee in Mansoura university (IRB number MD.21.01.404). Written Informed consent was obtained from the patients (if above 16) or patient’s legal guardian for participation in the research. All methods were performed in accordance with the ethical standards as laid down in the Declaration of Helsinki and its later amendments or comparable ethical standards. Consent for publication: Informed consent was obtained from the patient’s guardian for their child’s information to be published. Studies involving animals: Not applicable. Conflict of interest: No conflicts of interest or financial conflict.
Figures

Similar articles
-
The earliest enzyme replacement for infantile-onset Pompe disease in Japan.Pediatr Int. 2022 Jan;64(1):e15286. doi: 10.1111/ped.15286. Pediatr Int. 2022. PMID: 36074069
-
Use of cardiac magnetic resonance imaging to evaluate cardiac structure, function and fibrosis in children with infantile Pompe disease on enzyme replacement therapy.Mol Genet Metab. 2010 Dec;101(4):332-7. doi: 10.1016/j.ymgme.2010.07.011. Epub 2010 Jul 23. Mol Genet Metab. 2010. PMID: 20875764 Free PMC article.
-
Clinical course, mutations and its functional characteristics of infantile-onset Pompe disease in Thailand.BMC Med Genet. 2019 Sep 11;20(1):156. doi: 10.1186/s12881-019-0878-8. BMC Med Genet. 2019. PMID: 31510962 Free PMC article.
-
Pompe disease: early diagnosis and early treatment make a difference.Pediatr Neonatol. 2013 Aug;54(4):219-27. doi: 10.1016/j.pedneo.2013.03.009. Epub 2013 Apr 28. Pediatr Neonatol. 2013. PMID: 23632029 Review.
-
Expert Group Consensus on early diagnosis and management of infantile-onset pompe disease in the Gulf Region.Orphanet J Rare Dis. 2022 Oct 27;17(1):388. doi: 10.1186/s13023-022-02545-w. Orphanet J Rare Dis. 2022. PMID: 36303251 Free PMC article. Review.
References
-
- Wisselaar HA, et al. Structural and functional changes of lysosomal acid alpha-glucosidase during intracellular transport and maturation. J Biol Chem. 1993;268(3):2223–31. - PubMed
-
- Byrne BJ, et al. Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol Genet Metab. 2011;103(1):1–11. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
