

Derivation and Characterization of Isogenic OPA1 Mutant and Control Human Pluripotent Stem Cell Lines
- PMID: 39851566
- PMCID: PMC11764107
- DOI: 10.3390/cells14020137
Derivation and Characterization of Isogenic OPA1 Mutant and Control Human Pluripotent Stem Cell Lines
Abstract
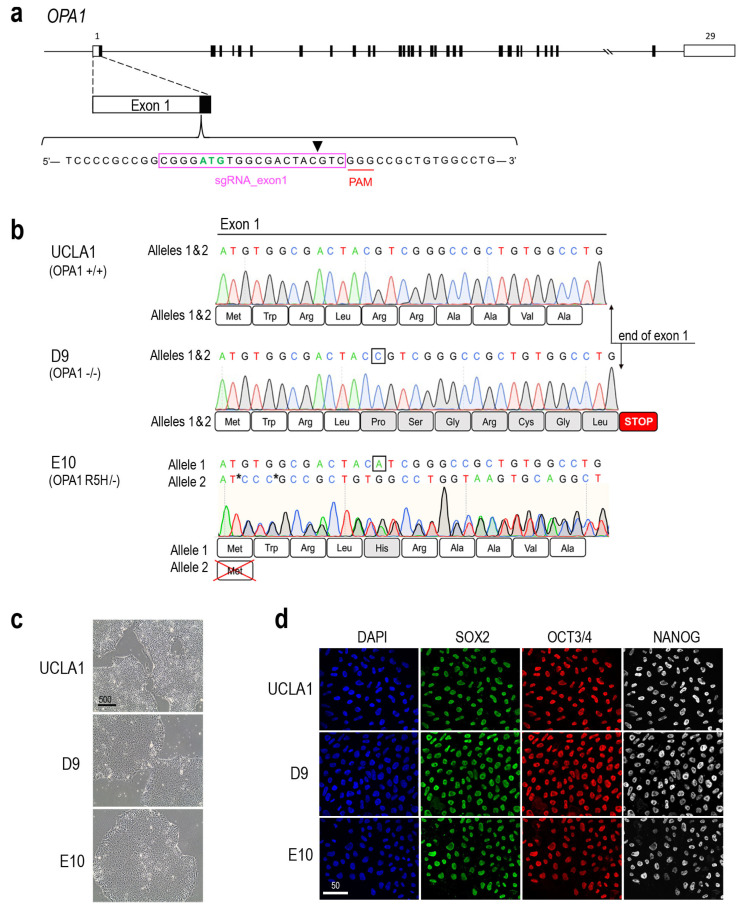
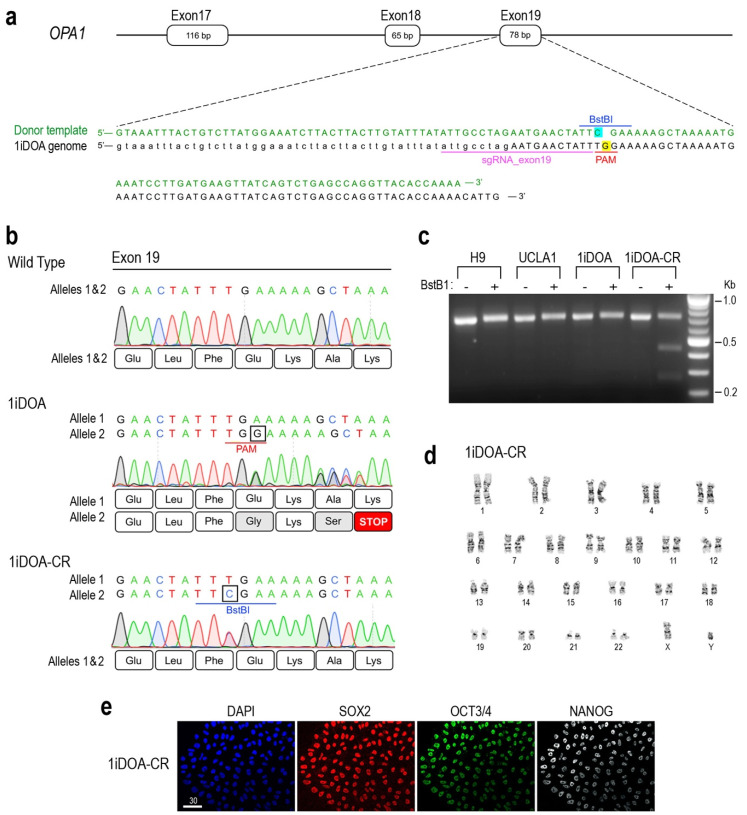
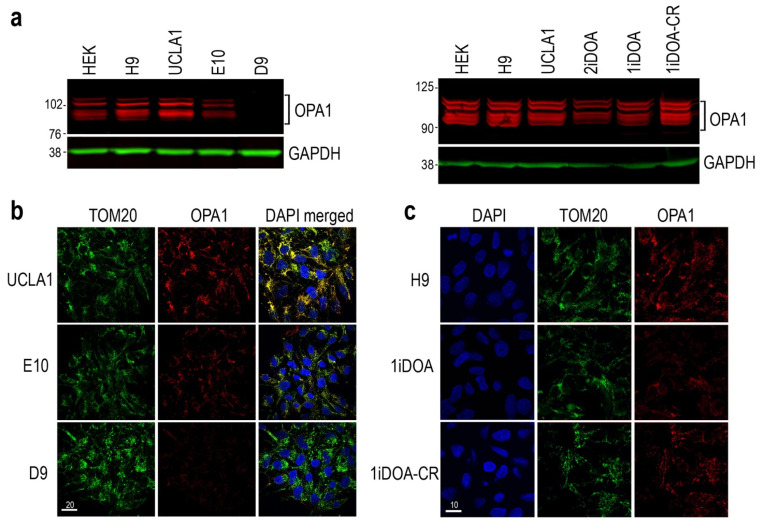
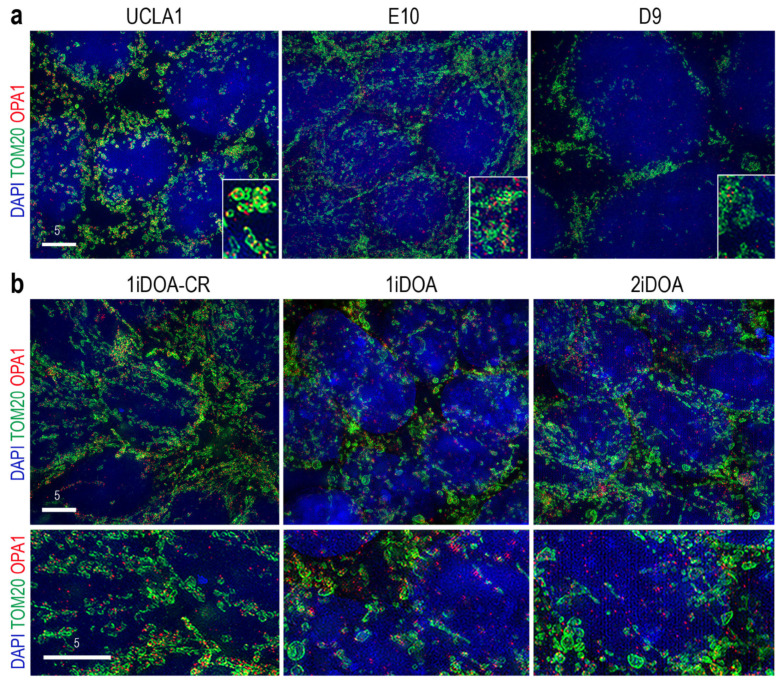
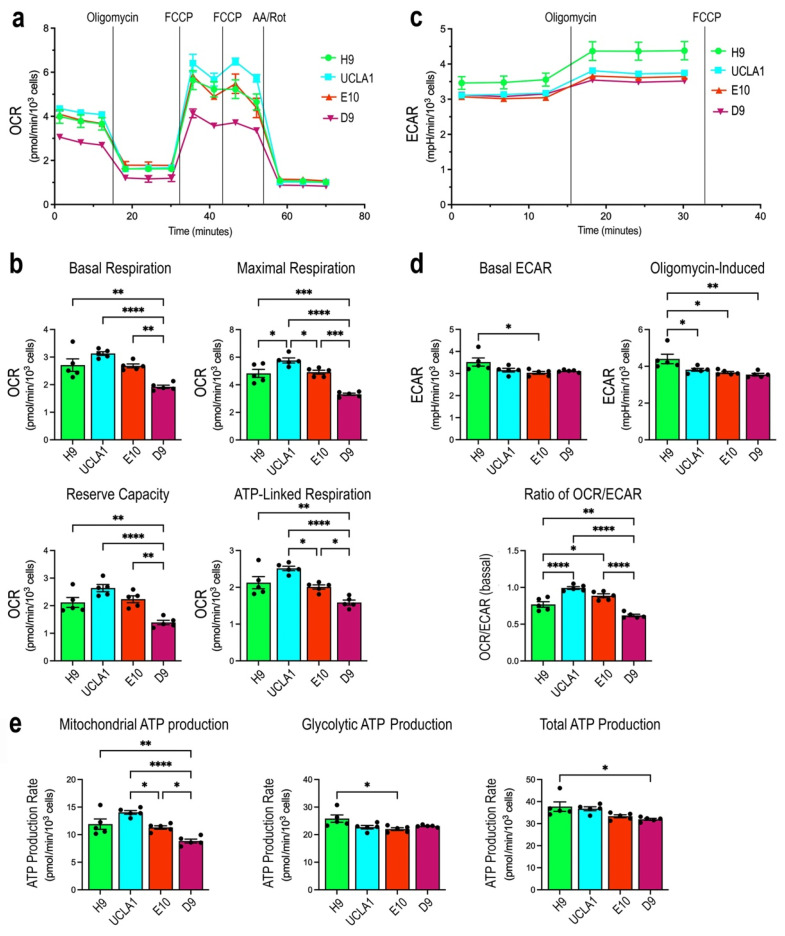
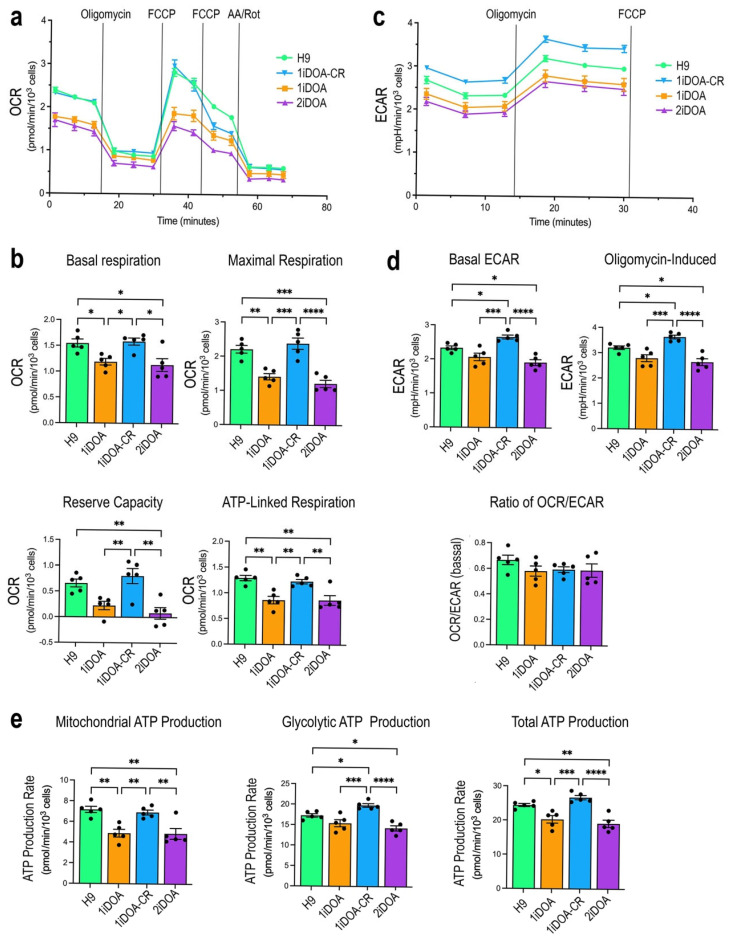
Dominant optic atrophy (DOA) is the most commonly inherited optic neuropathy. The majority of DOA is caused by mutations in the OPA1 gene, which encodes a dynamin-related GTPase located to the mitochondrion. OPA1 has been shown to regulate mitochondrial dynamics and promote fusion. Within the mitochondrion, proteolytically processed OPA1 proteins form complexes to maintain membrane integrity and the respiratory chain complexity. Although OPA1 is broadly expressed, human OPA1 mutations predominantly affect retinal ganglion cells (RGCs) that are responsible for transmitting visual information from the retina to the brain. Due to the scarcity of human RGCs, DOA has not been studied in depth using the disease affected neurons. To enable studies of DOA using stem-cell-derived human RGCs, we performed CRISPR-Cas9 gene editing to generate OPA1 mutant pluripotent stem cell (PSC) lines with corresponding isogenic controls. CRISPR-Cas9 gene editing yielded both OPA1 homozygous and heterozygous mutant ESC lines from a parental control ESC line. In addition, CRISPR-mediated homology-directed repair (HDR) successfully corrected the OPA1 mutation in a DOA patient's iPSCs. In comparison to the isogenic controls, the heterozygous mutant PSCs expressed the same OPA1 protein isoforms but at reduced levels; whereas the homozygous mutant PSCs showed a loss of OPA1 protein and altered mitochondrial morphology. Furthermore, OPA1 mutant PSCs exhibited reduced rates of oxygen consumption and ATP production associated with mitochondria. These isogenic PSC lines will be valuable tools for establishing OPA1-DOA disease models in vitro and developing treatments for mitochondrial deficiency associated neurodegeneration.
Keywords: CRISPR-Cas9 editing; OPA1 gene; dominant optic atrophy; isogenic human pluripotent stem cell lines; mitochondria.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Disruption of mitochondrial homeostasis and permeability transition pore opening in OPA1 iPSC-derived retinal ganglion cells.Acta Neuropathol Commun. 2025 Feb 13;13(1):28. doi: 10.1186/s40478-025-01942-z. Acta Neuropathol Commun. 2025. PMID: 39948685 Free PMC article.
-
CRISPRa-Mediated Increase of OPA1 Expression in Dominant Optic Atrophy.Int J Mol Sci. 2025 Jul 2;26(13):6364. doi: 10.3390/ijms26136364. Int J Mol Sci. 2025. PMID: 40650142 Free PMC article.
-
Modelling autosomal dominant optic atrophy associated with OPA1 variants in iPSC-derived retinal ganglion cells.Hum Mol Genet. 2022 Oct 10;31(20):3478-3493. doi: 10.1093/hmg/ddac128. Hum Mol Genet. 2022. PMID: 35652445 Free PMC article.
-
Dominant optic atrophy: Culprit mitochondria in the optic nerve.Prog Retin Eye Res. 2021 Jul;83:100935. doi: 10.1016/j.preteyeres.2020.100935. Epub 2020 Dec 17. Prog Retin Eye Res. 2021. PMID: 33340656 Review.
-
Interventions for central serous chorioretinopathy: a network meta-analysis.Cochrane Database Syst Rev. 2025 Jun 16;6(6):CD011841. doi: 10.1002/14651858.CD011841.pub3. Cochrane Database Syst Rev. 2025. PMID: 40522203
References
-
- Yu-Wai-Man P., Griffiths P.G., Burke A., Sellar P.W., Clarke M.P., Gnanaraj L., Ah-Kine D., Hudson G., Czermin B., Taylor R.W., et al. The prevalence and natural history of dominant optic atrophy due to OPA1 mutations. Ophthalmology. 2010;117:1538–1546.e1. doi: 10.1016/j.ophtha.2009.12.038. - DOI - PMC - PubMed
-
- Kjer P. Infantile optic atrophy with dominant mode of inheritance: A clinical and genetic study of 19 Danish families. Acta Ophthalmol. Suppl. 1959;164:1–147. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
