

Metabolic dependency mapping identifies Peroxiredoxin 1 as a driver of resistance to ATM inhibition
- PMID: 39854937
- PMCID: PMC11795153
- DOI: 10.1016/j.redox.2025.103503
Metabolic dependency mapping identifies Peroxiredoxin 1 as a driver of resistance to ATM inhibition
Abstract
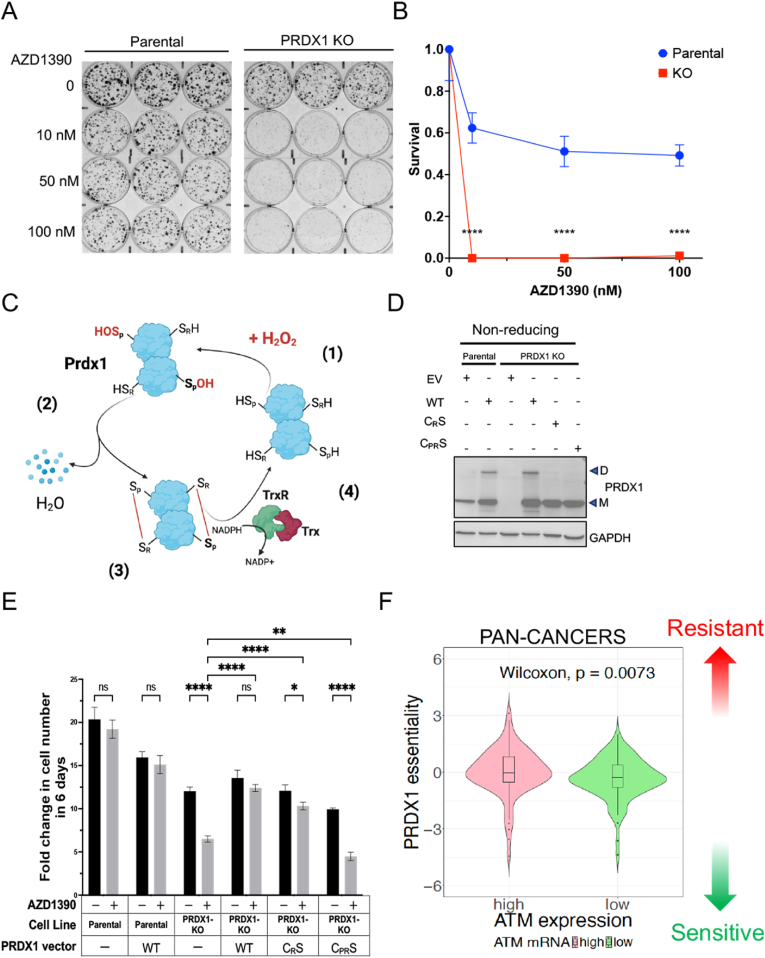
Metabolic pathways fuel tumor progression and resistance to stress conditions including chemotherapeutic drugs, such as DNA damage response (DDR) inhibitors. Yet, significant gaps persist in how metabolic pathways confer resistance to DDR inhibition in cancer cells. Here, we employed a metabolism-focused CRISPR knockout screen and identified genetic vulnerabilities to DDR inhibitors. We unveiled Peroxiredoxin 1 (PRDX1) as a synthetic lethality partner with Ataxia Telangiectasia Mutated (ATM) kinase. Tumor cells depleted of PRDX1 displayed heightened sensitivity to ATM inhibition in vitro and in mice in a manner dependent on p53 status. Mechanistically, we discovered that the ribosomal protein RPL32 undergoes redox modification on active cysteine residues 91 and 96 upon ATM inhibition, promoting p53 stability and altered cell fitness. Our findings reveal a new pathway whereby RPL32 senses stress and induces p53 activation impairing tumor cell survival.
Keywords: ATM kinase; Disulfide stress; Peroxiredoxin 1; RPL32 redox modification; p53 activation.
Published by Elsevier B.V.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures








References
-
- Blackford A.N., Jackson S.P. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell. 2017;66:801–817. - PubMed
-
- Shiloh Y., Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013;14:197–210. - PubMed
-
- Lee J.H., Paull T.T. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat. Rev. Mol. Cell Biol. 2021;22:796–814. - PubMed
-
- Choi M., Kipps T., Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol. Cancer Therapeut. 2016;15:1781–1791. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
