

SLC25A38 is required for mitochondrial pyridoxal 5'-phosphate (PLP) accumulation
- PMID: 39856062
- PMCID: PMC11760969
- DOI: 10.1038/s41467-025-56130-3
SLC25A38 is required for mitochondrial pyridoxal 5'-phosphate (PLP) accumulation
Abstract
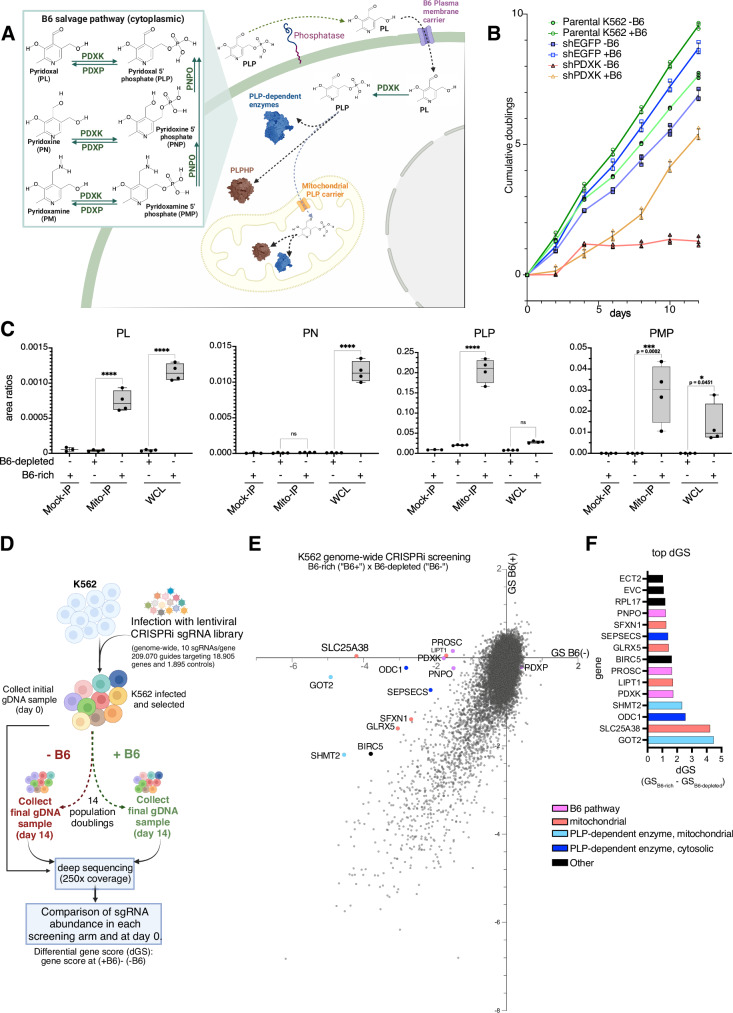
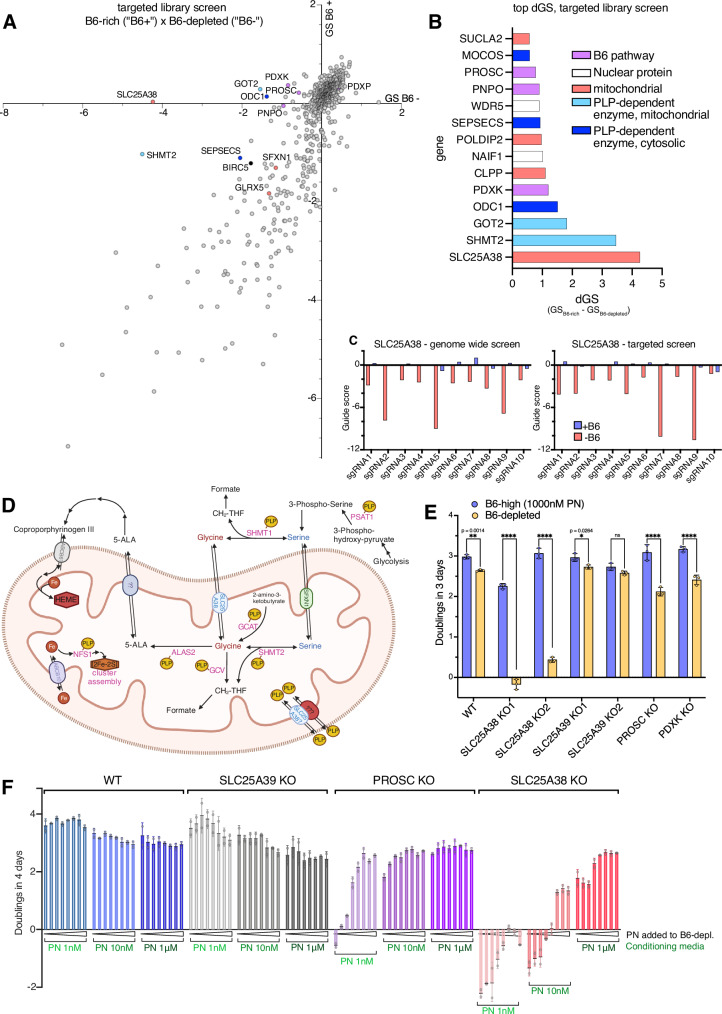
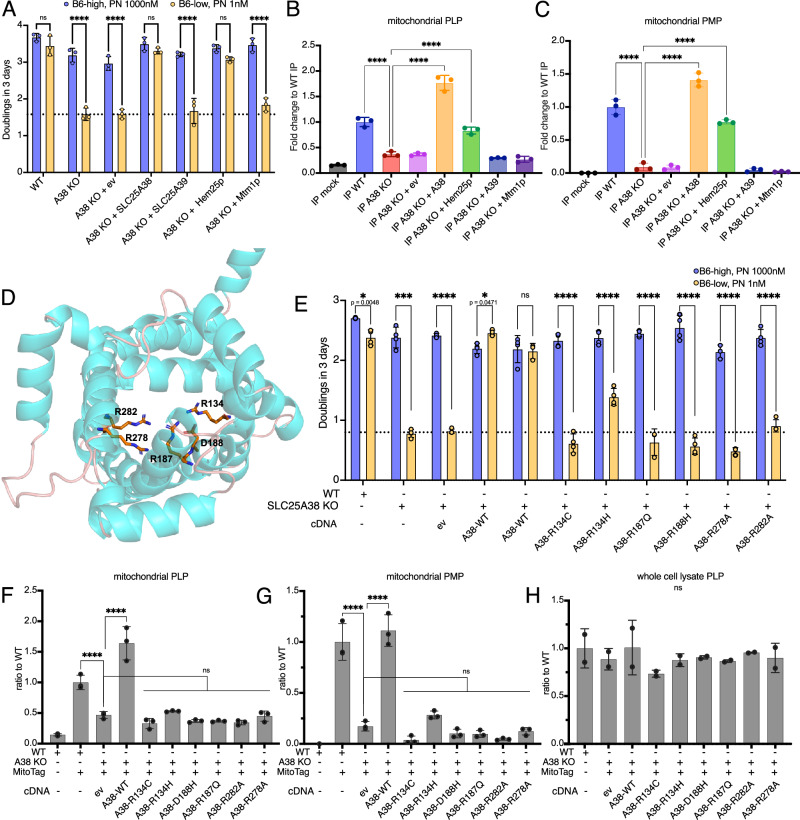
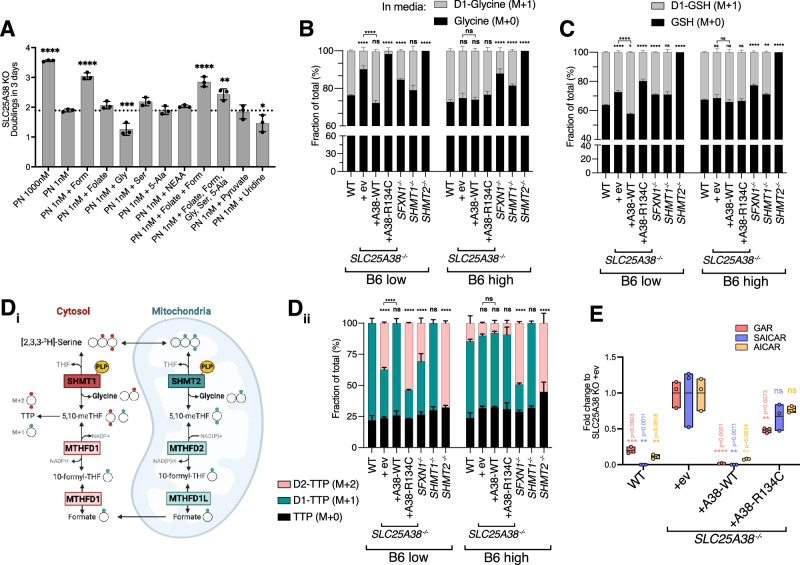
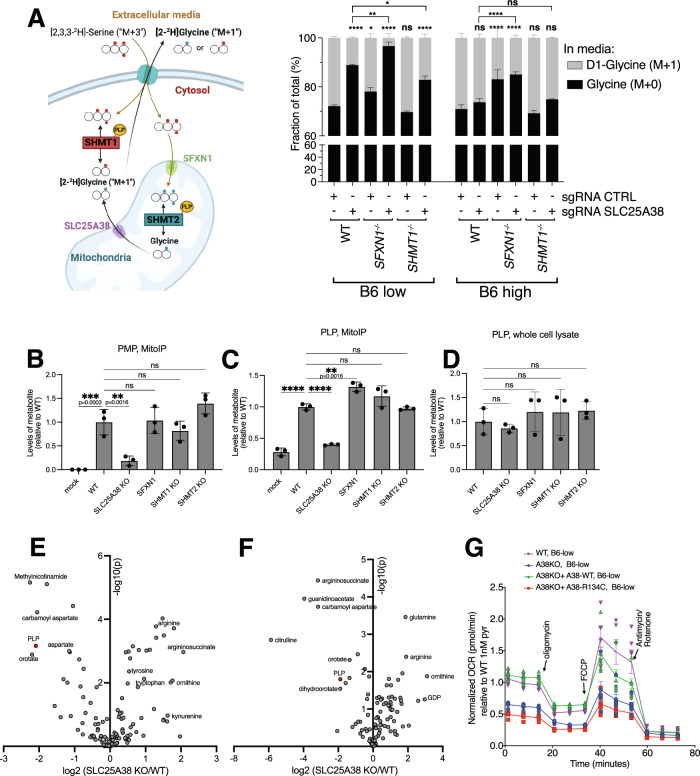
Many essential proteins require pyridoxal 5'-phosphate, the active form of vitamin B6, as a cofactor for their activity. These include enzymes important for amino acid metabolism, one-carbon metabolism, polyamine synthesis, erythropoiesis, and neurotransmitter metabolism. A third of all mammalian pyridoxal 5'-phosphate-dependent enzymes are localized in the mitochondria; however, the molecular machinery involved in the regulation of mitochondrial pyridoxal 5'-phosphate levels in mammals remains unknown. In this study, we used a genome-wide CRISPR interference screen in erythroleukemia cells and organellar metabolomics to identify the mitochondrial inner membrane protein SLC25A38 as a regulator of mitochondrial pyridoxal 5'-phosphate. Loss of SLC25A38 causes depletion of mitochondrial, but not cellular, pyridoxal 5'-phosphate, and impairs cellular proliferation under both physiological and low vitamin B6 conditions. Metabolic changes associated with SLC25A38 loss suggest impaired mitochondrial pyridoxal 5'-phosphate-dependent enzymatic reactions, including serine to glycine conversion catalyzed by serine hydroxymethyltransferase-2 as well as ornithine aminotransferase. The proliferation defect of SLC25A38-null K562 cells in physiological and low vitamin B6 media can be explained by the loss of serine hydroxymethyltransferase-2-dependent production of one-carbon units and downstream de novo nucleotide synthesis. Our work points to a role for SLC25A38 in mitochondrial pyridoxal 5'-phosphate accumulation and provides insights into the pathology of congenital sideroblastic anemia.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: M.G.V.H. discloses that he is an advisor for Agios Pharmaceuticals, iTeos Therapeutics, Sage Therapeutics, Pretzel Therapeutics, Droia Ventures, MPM Capital, and Auron Therapeutics. G.S. is currently an employee and shareholder of AstraZeneca. All other authors declare that they have no competing interests.
Figures

References
-
- Wilson, M. P., Plecko, B., Mills, P. B. & Clayton, P. T. Disorders affecting vitamin B6 metabolism. J. Inherit. Metab. Dis.42, 629–646 (2019). - PubMed
-
- van Karnebeek, C., Pena, I. A. & Gospe, S. M. Disorders of pyridoxine metabolism. In (Rosenberg, R. N. & Pascual, J. M. eds.) Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease (Sixth Edition) 711–728 (Academic Press, 2020).
-
- Waymire, K. G. et al. Mice lacking tissue non–specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B–6. Nat. Genet.11, 45–51 (1995). - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
- P30 CA014051/CA/NCI NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- P30 DA018343/DA/NIDA NIH HHS/United States
- R35 CA242379/CA/NCI NIH HHS/United States
- F31 CA228241/CA/NCI NIH HHS/United States
- T32 GM144273/GM/NIGMS NIH HHS/United States
- R35 GM151097/GM/NIGMS NIH HHS/United States
- R00 CA241332/CA/NCI NIH HHS/United States
- F31 NS127458/NS/NINDS NIH HHS/United States
- T32 GM007287/GM/NIGMS NIH HHS/United States
- F30 CA268633/CA/NCI NIH HHS/United States
- T32 HL007118/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
