

Genotype-phenotype correlations for 17 Chinese families with inherited retinal dystrophies due to homozygous variants
- PMID: 39856360
- PMCID: PMC11759671
- DOI: 10.1038/s41598-025-87844-5
Genotype-phenotype correlations for 17 Chinese families with inherited retinal dystrophies due to homozygous variants
Abstract
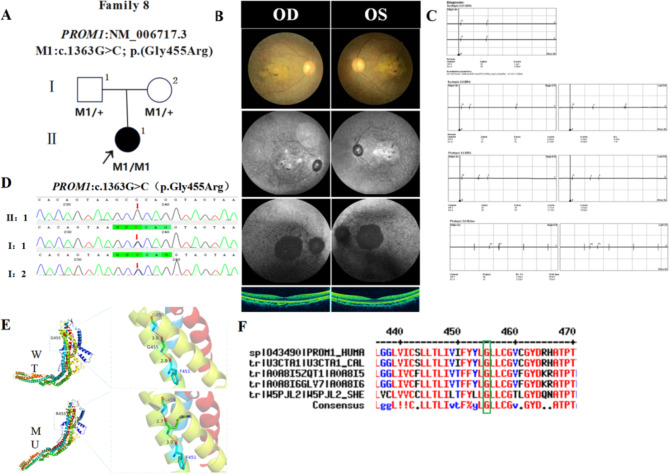
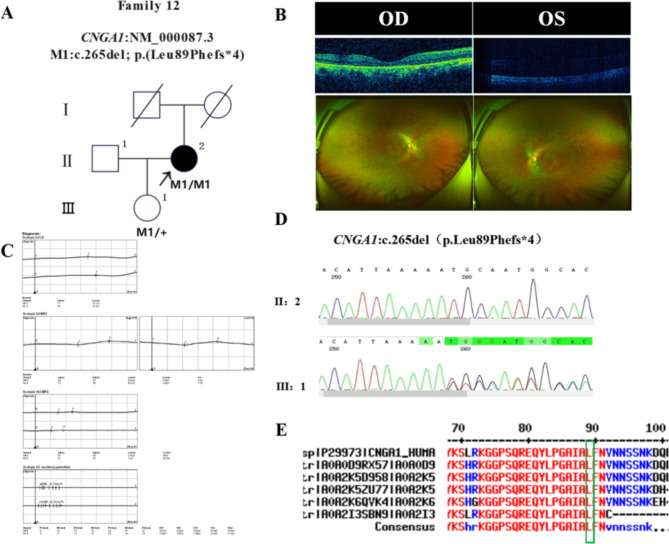
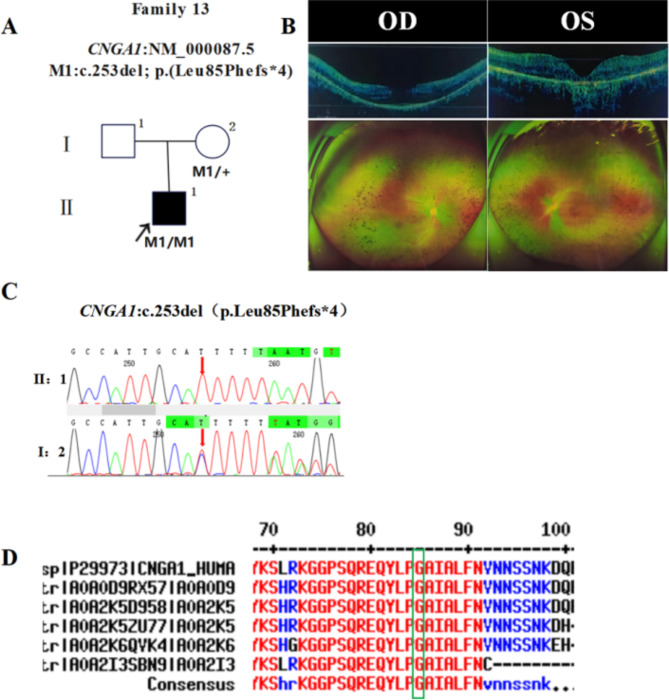
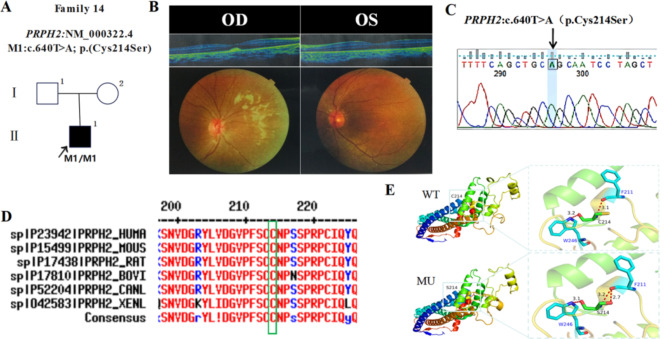
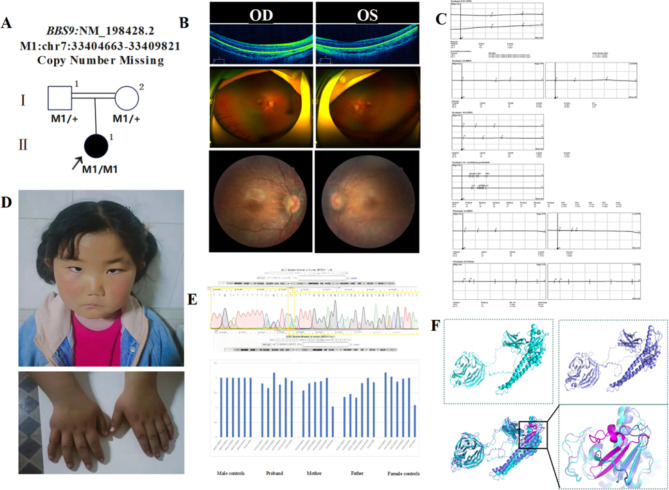
In this study, patients with inherited retinal dystrophies (IRDs) who visited Ningxia Eye Hospital from January 2015 to September 2023 were analyzed. Through Whole Exome Sequencing (WES) and Sanger verification, 17 probands carrying homozygous variants were detected. The association between the genotype and clinical phenotype of patients with homozygous variants was analyzed. Among all the patients, 3 patients (17.6%) had a family history of consanguineous marriage, and the onset age of 5 patients(29.4%) was less than 10 years. According to 12 patients (70.6% ), they had the best corrected visual acuity (monocular) < 0.3. 3 were blind, 9 with moderate to severe visual impairment, and 2 with mild visual impairment. 16 homozygous variants were detected in 9 different genes, of which 7 were novel homozygous variants, including frameshift variants, missense variants, and a copy number variant. These variants are related to clinical phenotypes such as Usher syndrome type II (USH2), Stargardt disease (STGD), retinitis pigmentosa (RP), Leber congenital amaurosis (LCA), and Bardet-Biedl syndrome (BBS) respectively. The results of the study indicate that more than 80% of persons with homozygous variant originated from non-consanguineous families, emphasizing the significance of genetic screening for individuals who lack a family history of consanguineous marriage and no obvious clinical phenotypes, but who may carry genetic pathogenic variants for genetic diseases. Furthermore, by analyzing the genotypes and clinical phenotypes of IRD patients from these 17 Chinese families, we have expanded the spectrum of variants in known pathogenic genes for IRDs and the range of clinical phenotypes associated with variants in these genes. We have identified couples at high risk of having affected offspring and individuals with moderate to severe IRDs, providing a basis for genetic counseling, reproductive decision-making, disease prevention, and management. Our findings highlight the association between homozygous variants and more severe clinical phenotypes within these families, thus laying the groundwork for future genetic screening and intervention strategies.
Keywords: Clinical phenotype; Consanguineous marriage; Genotype; Homozygous variation; Inherited retinal dystrophies.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests. Ethics approval and consent to participate: The Ethics Committee on Human Research at People Hospital in the Ningxia Hui Autonomous Region accepted and examined our work (reference number: 2022-KJCG-006), which adhered to the Declaration of Helsinki. Each participant or their legal guardians provided their written informed permission before to taking part.
Figures


Similar articles
-
Genotypic profile and phenotype correlations of ABCA4-associated retinopathy in Koreans.Mol Vis. 2019 Nov 14;25:679-690. eCollection 2019. Mol Vis. 2019. PMID: 31814693 Free PMC article.
-
Screening of a large cohort of leber congenital amaurosis and retinitis pigmentosa patients identifies novel LCA5 mutations and new genotype-phenotype correlations.Hum Mutat. 2013 Nov;34(11):1537-1546. doi: 10.1002/humu.22398. Epub 2013 Sep 17. Hum Mutat. 2013. PMID: 23946133 Free PMC article.
-
Deciphering the Genetic Basis of Degenerative and Developmental Eye Disorders in 50 Pakistani Consanguineous Families Using Whole-Exome Sequencing.Int J Mol Sci. 2025 Mar 18;26(6):2715. doi: 10.3390/ijms26062715. Int J Mol Sci. 2025. PMID: 40141357 Free PMC article.
-
Gene Therapy in Hereditary Retinal Dystrophies: The Usefulness of Diagnostic Tools in Candidate Patient Selections.Int J Mol Sci. 2023 Sep 6;24(18):13756. doi: 10.3390/ijms241813756. Int J Mol Sci. 2023. PMID: 37762059 Free PMC article. Review.
-
A systematic review of inherited retinal dystrophies in Pakistan: updates from 1999 to April 2023.BMC Ophthalmol. 2024 Feb 5;24(1):55. doi: 10.1186/s12886-024-03319-7. BMC Ophthalmol. 2024. PMID: 38317096 Free PMC article.
References
Publication types
MeSH terms
Supplementary concepts
Grants and funding
- 2024AAC03536/National Natural Science Foundation of Ningxia Hui Autonomous Region
- 82260206/National Natural Science Foundation of China
- 2024BEG02017/key research development project of Ningxia Hui Autonomous Region
- 2020GKLRLX13/the training project of the scientific innovation commanding talented person in Ningxia Hui Autonomous Region
- 2022CJE09011/Major achievement transformation project of Ningxia Hui Autonomous Region
LinkOut - more resources
Full Text Sources
Research Materials
