

Longitudinal phage-bacteria dynamics in the early life gut microbiome
- PMID: 39856391
- PMCID: PMC11790489
- DOI: 10.1038/s41564-024-01906-4
Longitudinal phage-bacteria dynamics in the early life gut microbiome
Abstract
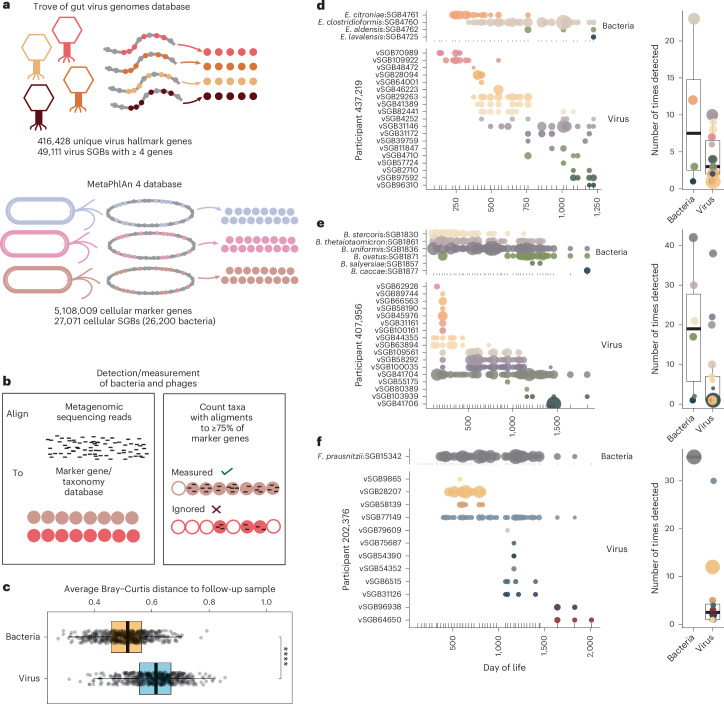
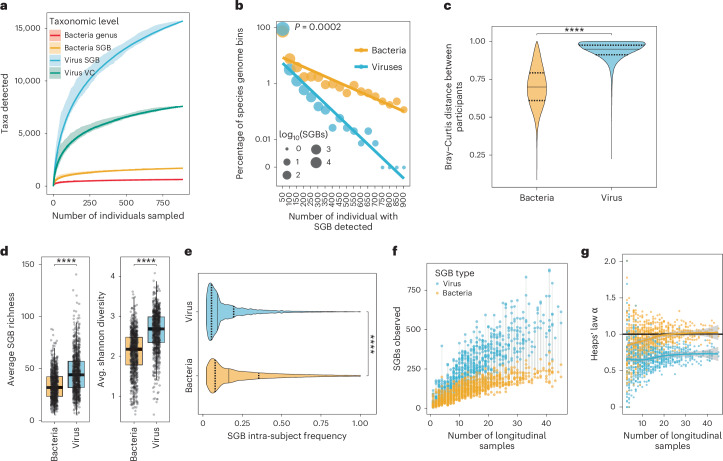
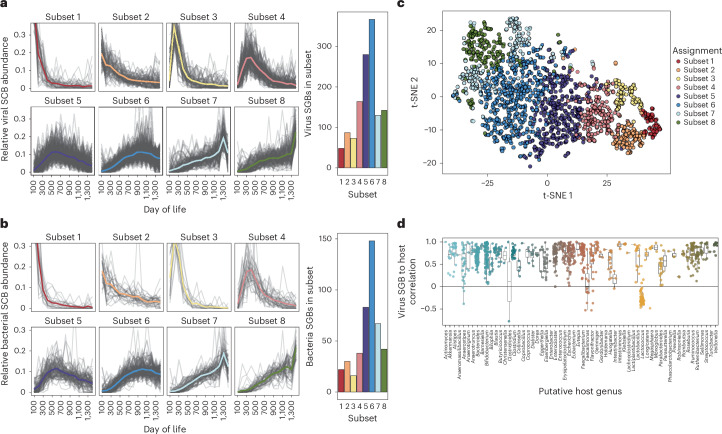
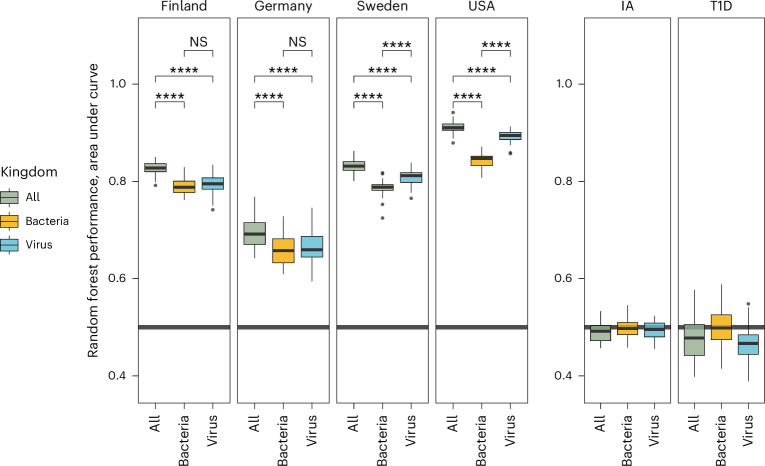
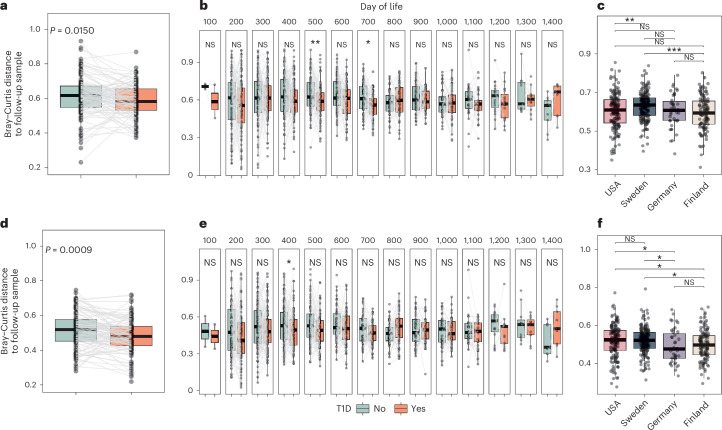
Microbial colonization of the human gut occurs soon after birth, proceeds through well-studied phases and is affected by lifestyle and other factors. Less is known about phage community dynamics during infant gut colonization due to small study sizes, an inability to leverage large databases and a lack of appropriate bioinformatics tools. Here we reanalysed whole microbial community shotgun sequencing data of 12,262 longitudinal samples from 887 children from four countries across four years of life as part of the The Environmental Determinants of Diabetes in the Young (TEDDY) study. We developed an extensive metagenome-assembled genome catalogue using the Marker-MAGu pipeline, which comprised 49,111 phage taxa from existing human microbiome datasets. This was used to identify phage marker genes and their integration into the MetaPhlAn 4 bacterial marker gene database enabled simultaneous assessment of phage and bacterial dynamics. We found that individual children are colonized by hundreds of different phages, which are more transitory than bacteria, accumulating a more diverse phage community over time. Type 1 diabetes correlated with a decreased rate of change in bacterial and viral communities in children aged one and two. The addition of phage data improved the ability of machine learning models to discriminate samples by country. Finally, although phage populations were specific to individuals, we observed trends of phage ecological succession that correlated well with putative host bacteria. This resource improves our understanding of phage-bacteria interactions in the developing early life microbiome.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures









References
-
- Nieuwdorp, M., Gilijamse, P. W., Pai, N. & Kaplan, L. M. Role of the microbiome in energy regulation and metabolism. Gastroenterology146, 1525–1533 (2014). - PubMed
-
- Jameson, K. G., Olson, C. A., Kazmi, S. A. & Hsiao, E. Y. Toward understanding microbiome–neuronal signaling. Mol. Cell78, 577–583 (2020). - PubMed
MeSH terms
Grants and funding
- U01 DK063821/DK/NIDDK NIH HHS/United States
- UC4 DK063863/DK/NIDDK NIH HHS/United States
- UL1 TR002535/TR/NCATS NIH HHS/United States
- U01 DK063790/DK/NIDDK NIH HHS/United States
- UL1 TR000064/TR/NCATS NIH HHS/United States
- HHSN267200700014C/LM/NLM NIH HHS/United States
- U01 DK063836/DK/NIDDK NIH HHS/United States
- U01 DK063829/DK/NIDDK NIH HHS/United States
- U01 DK063865/DK/NIDDK NIH HHS/United States
- UC4 DK095300/DK/NIDDK NIH HHS/United States
- U01 DK63865/U.S. Department of Health & Human Services | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (National Institute of Diabetes & Digestive & Kidney Diseases)
- UC4 DK063861/DK/NIDDK NIH HHS/United States
- UC4 DK063829/DK/NIDDK NIH HHS/United States
- UC4 DK063821/DK/NIDDK NIH HHS/United States
- U01 DK63821/U.S. Department of Health & Human Services | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (National Institute of Diabetes & Digestive & Kidney Diseases)
- UC4 DK117483/DK/NIDDK NIH HHS/United States
- UC4 DK063836/DK/NIDDK NIH HHS/United States
- UC4 DK112243/DK/NIDDK NIH HHS/United States
- U01 DK124166/DK/NIDDK NIH HHS/United States
- U01 DK063861/DK/NIDDK NIH HHS/United States
- P30 ES030285/ES/NIEHS NIH HHS/United States
- U01 DK63829/U.S. Department of Health & Human Services | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (National Institute of Diabetes & Digestive & Kidney Diseases)
- U01 DK128847/DK/NIDDK NIH HHS/United States
- UC4 DK063865/DK/NIDDK NIH HHS/United States
- U01 DK063863/DK/NIDDK NIH HHS/United States
- UC4 DK106955/DK/NIDDK NIH HHS/United States
- UC4 DK100238/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
