

TRIM28-dependent developmental heterogeneity determines cancer susceptibility through distinct epigenetic states
- PMID: 39856421
- PMCID: PMC11864977
- DOI: 10.1038/s43018-024-00900-3
TRIM28-dependent developmental heterogeneity determines cancer susceptibility through distinct epigenetic states
Abstract
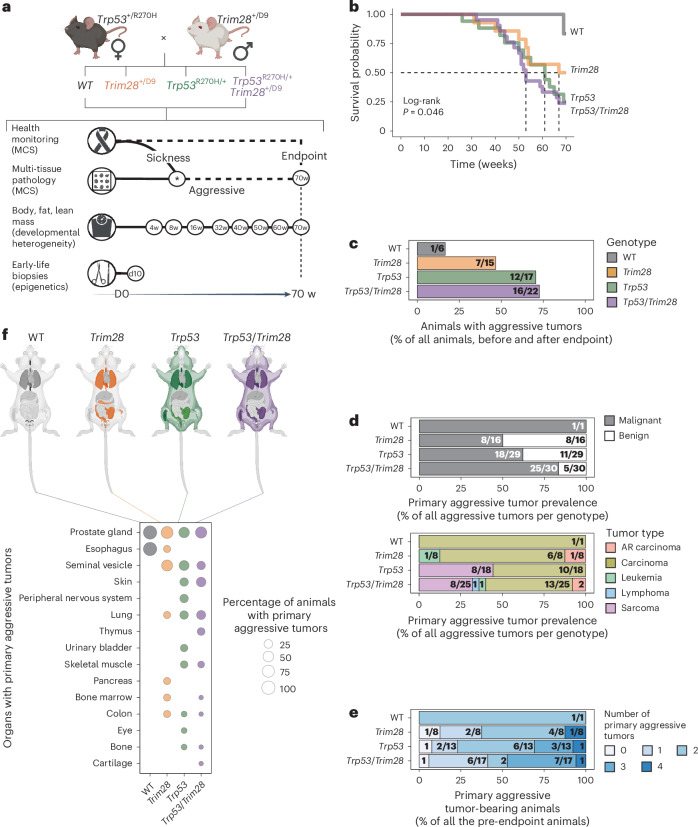
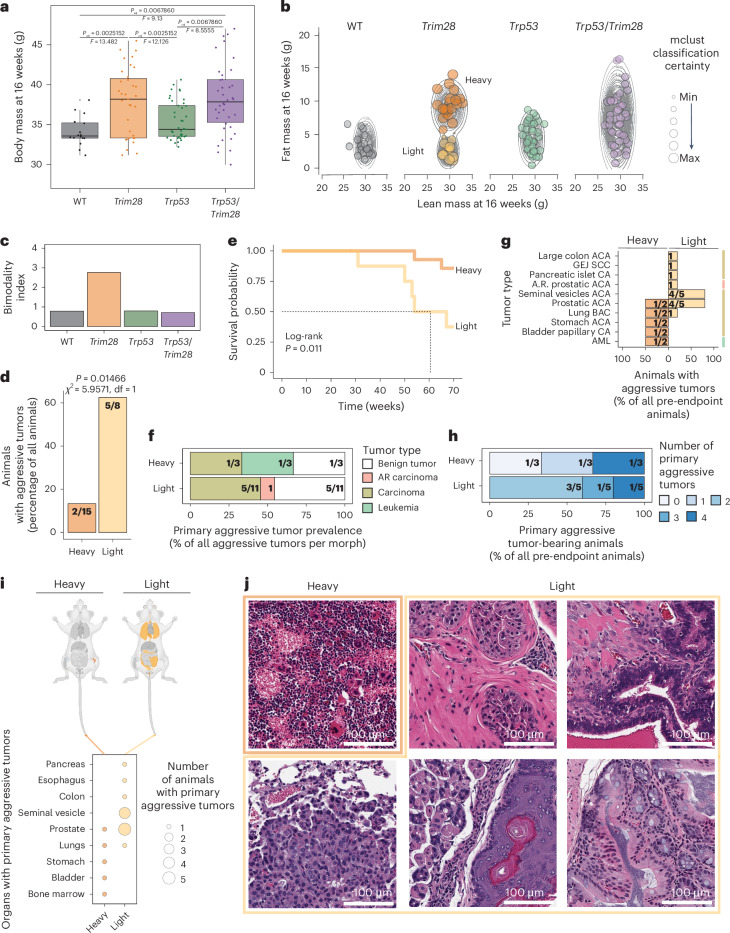
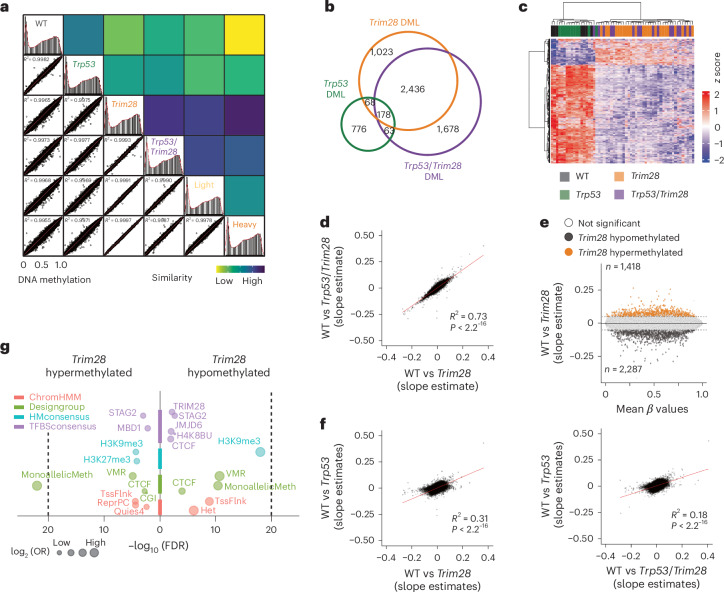
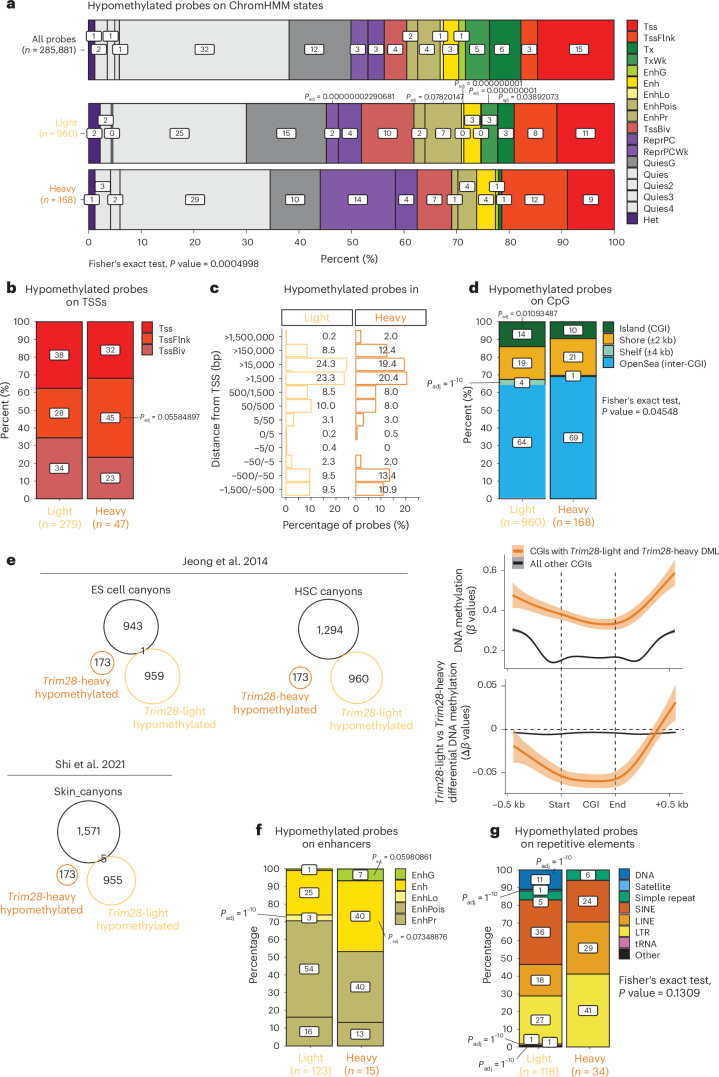
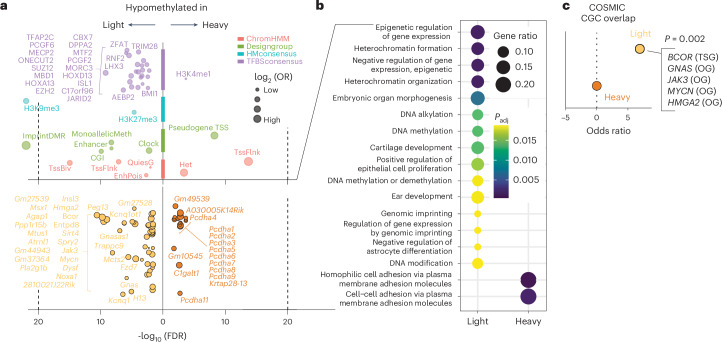
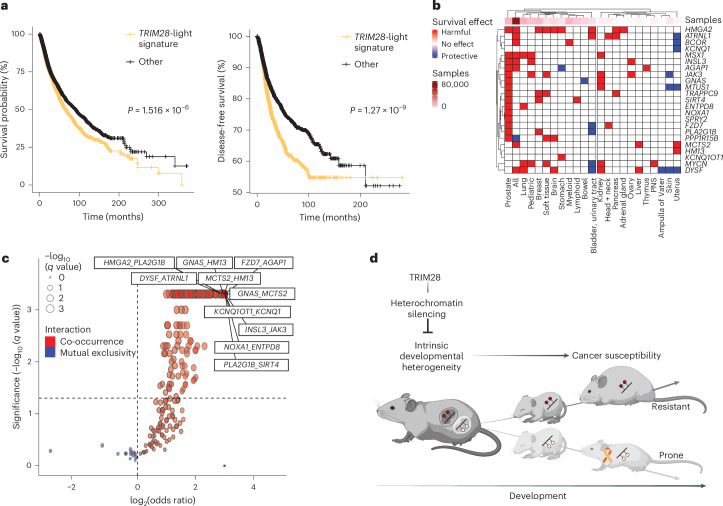
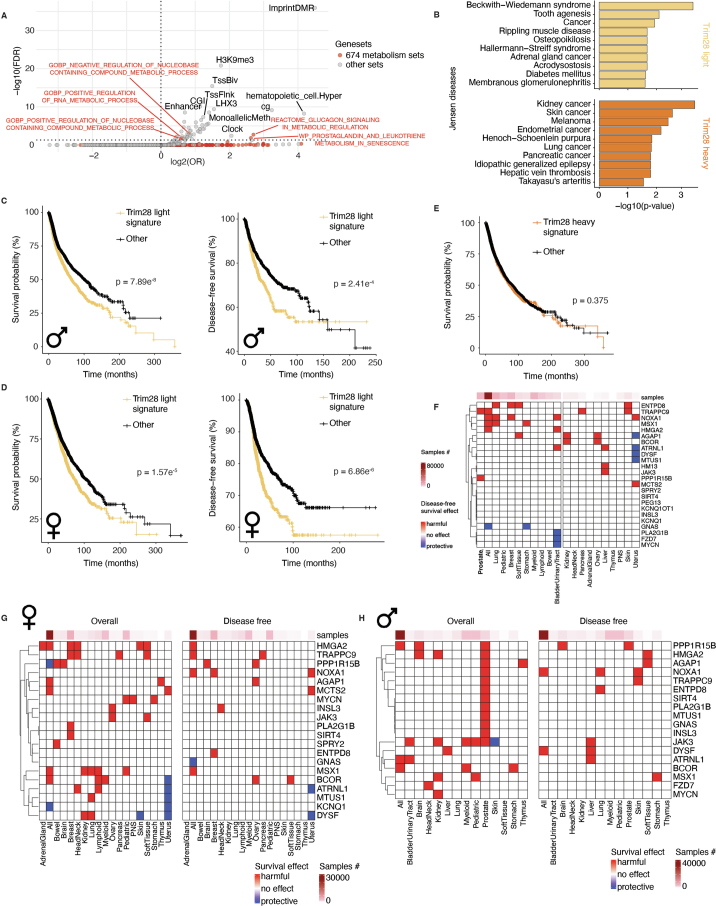
Mutations in cancer risk genes increase susceptibility, but not all carriers develop cancer. Indeed, while DNA mutations are necessary drivers of cancer, only a small subset of mutated cells go on to cause the disease. To date, the mechanisms underlying individual cancer susceptibility remain unclear. Here, we took advantage of a unique mouse model of intrinsic developmental heterogeneity (Trim28+/D9) to investigate whether early-life epigenetic variation influences cancer susceptibility later in life. We found that heterozygosity of Trim28 is sufficient to generate two distinct early-life epigenetic states associated with differing cancer susceptibility. These developmentally primed states exhibit differential methylation patterns at typically silenced heterochromatin, detectable as early as 10 days of age. The differentially methylated loci are enriched for genes with known oncogenic potential, frequently mutated in human cancers and correlated with poor prognosis. This study provides genetic evidence that intrinsic developmental heterogeneity can prime individual, lifelong cancer susceptibility.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures








Update of
-
Developmental priming of cancer susceptibility.bioRxiv [Preprint]. 2023 Sep 15:2023.09.12.557446. doi: 10.1101/2023.09.12.557446. bioRxiv. 2023. Update in: Nat Cancer. 2025 Feb;6(2):385-403. doi: 10.1038/s43018-024-00900-3. PMID: 37745326 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
