

Advancing long-read nanopore genome assembly and accurate variant calling for rare disease detection
- PMID: 39862869
- PMCID: PMC11866955
- DOI: 10.1016/j.ajhg.2025.01.002
Advancing long-read nanopore genome assembly and accurate variant calling for rare disease detection
Abstract
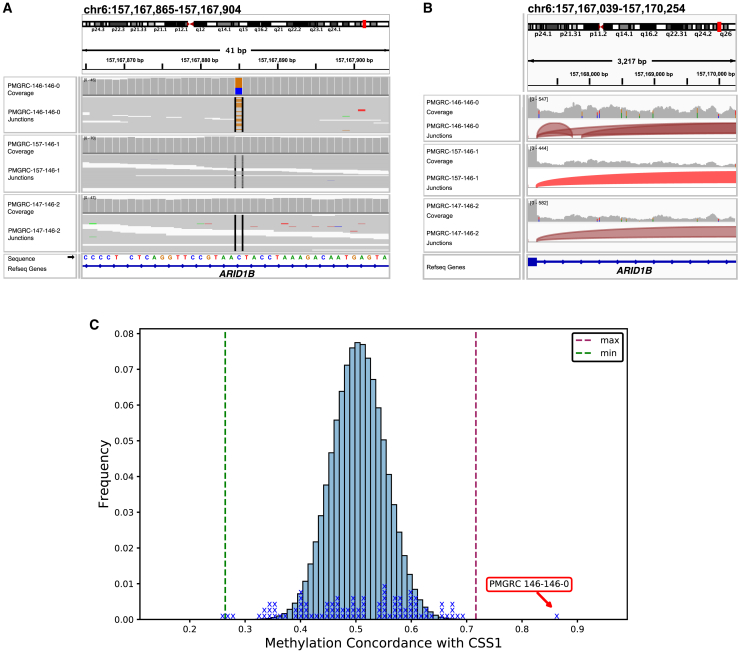
More than 50% of families with suspected rare monogenic diseases remain unsolved after whole-genome analysis by short-read sequencing (SRS). Long-read sequencing (LRS) could help bridge this diagnostic gap by capturing variants inaccessible to SRS, facilitating long-range mapping and phasing and providing haplotype-resolved methylation profiling. To evaluate LRS's additional diagnostic yield, we sequenced a rare-disease cohort of 98 samples from 41 families, using nanopore sequencing, achieving per sample ∼36× average coverage and 32-kb read N50 from a single flow cell. Our Napu pipeline generated assemblies, phased variants, and methylation calls. LRS covered, on average, coding exons in ∼280 genes and ∼5 known Mendelian disease-associated genes that were not covered by SRS. In comparison to SRS, LRS detected additional rare, functionally annotated variants, including structural variants (SVs) and tandem repeats, and completely phased 87% of protein-coding genes. LRS detected additional de novo variants and could be used to distinguish postzygotic mosaic variants from prezygotic de novos. Diagnostic variants were established by LRS in 11 probands, with diverse underlying genetic causes including de novo and compound heterozygous variants, large-scale SVs, and epigenetic modifications. Our study demonstrates LRS's potential to enhance diagnostic yield for rare monogenic diseases, implying utility in future clinical genomics workflows.
Keywords: clinical testing; gene conversion; haplotype phasing; long-read sequencing; methylation; rare-disease diagnosis; structural variants; variant annotation.
Copyright © 2025. Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests A.O.-L. was a paid consultant for Tome Biosciences, Ono Pharma USA, and Addition Therapeutics. The Rare Genomes Project received support in the form of reagents from Illumina Inc. and Pacific Biosciences.
Figures






Update of
-
Advancing long-read nanopore genome assembly and accurate variant calling for rare disease detection.medRxiv [Preprint]. 2024 Aug 22:2024.08.22.24312327. doi: 10.1101/2024.08.22.24312327. medRxiv. 2024. Update in: Am J Hum Genet. 2025 Feb 06;112(2):428-449. doi: 10.1016/j.ajhg.2025.01.002. PMID: 39228712 Free PMC article. Updated. Preprint.
References
-
- Kingsmore S.F., Cakici J.A., Clark M.M., Gaughran M., Feddock M., Batalov S., Bainbridge M.N., Carroll J., Caylor S.A., Clarke C., et al. A Randomized, Controlled Trial of the Analytic and Diagnostic Performance of Singleton and Trio, Rapid Genome and Exome Sequencing in Ill Infants. Am. J. Hum. Genet. 2019;105:719–733. doi: 10.1016/j.ajhg.2019.08.009. - DOI - PMC - PubMed
-
- Wojcik M.H., Reuter C.M., Marwaha S., Mahmoud M., Duyzend M.H., Barseghyan H., Yuan B., Boone P.M., Groopman E.E., Délot E.C., et al. Beyond the exome: What’s next in diagnostic testing for Mendelian conditions. Am. J. Hum. Genet. 2023;110:1229–1248. doi: 10.1016/j.ajhg.2023.06.009. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
