

Statistical identification of cell type-specific spatially variable genes in spatial transcriptomics
- PMID: 39865128
- PMCID: PMC11770176
- DOI: 10.1038/s41467-025-56280-4
Statistical identification of cell type-specific spatially variable genes in spatial transcriptomics
Abstract
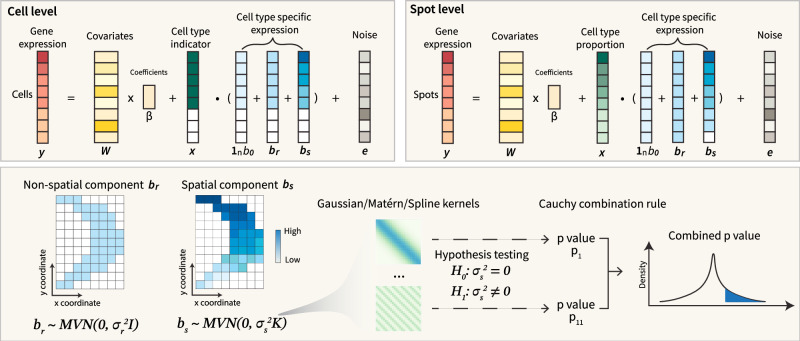
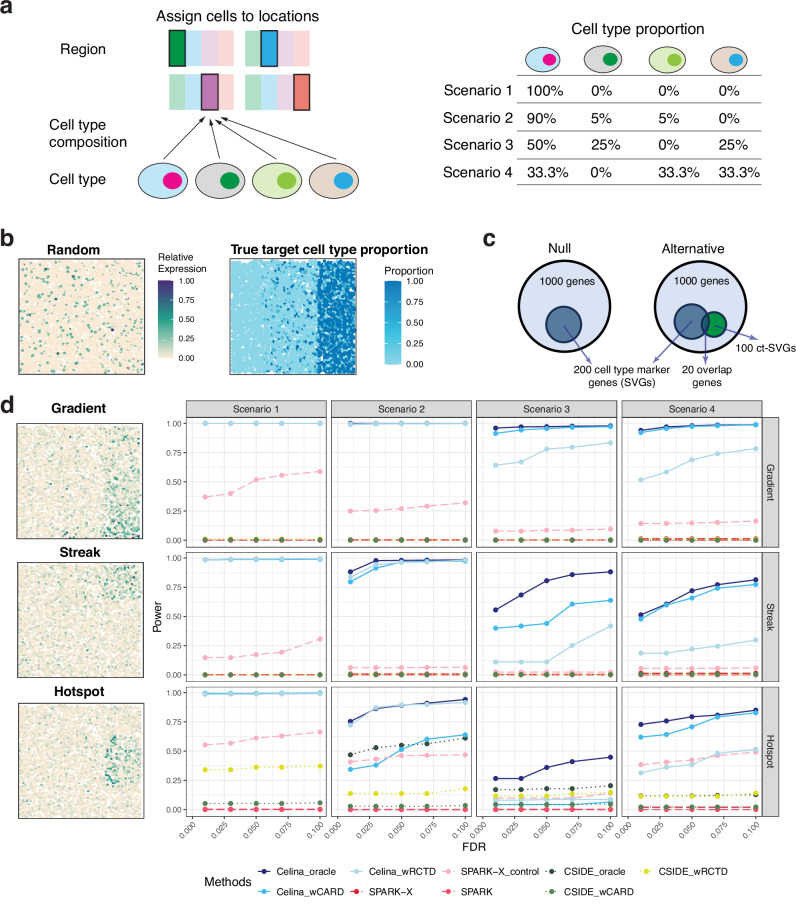
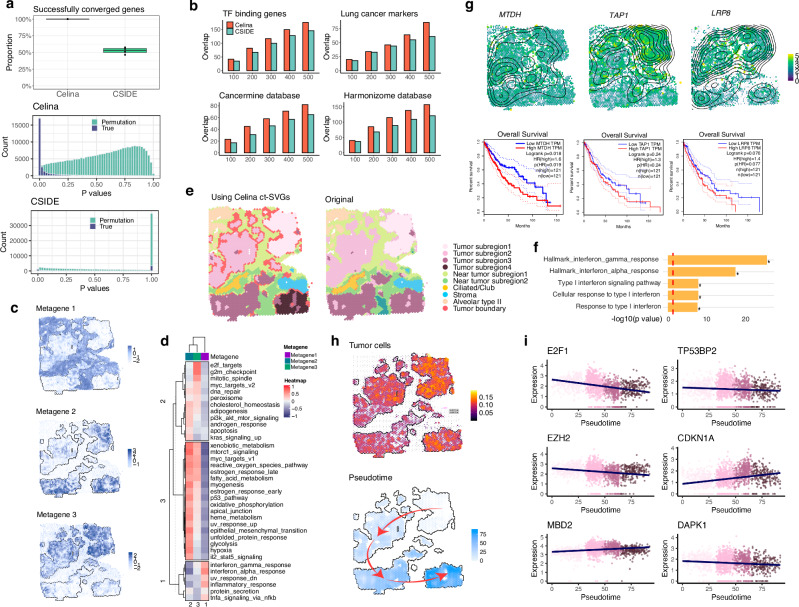
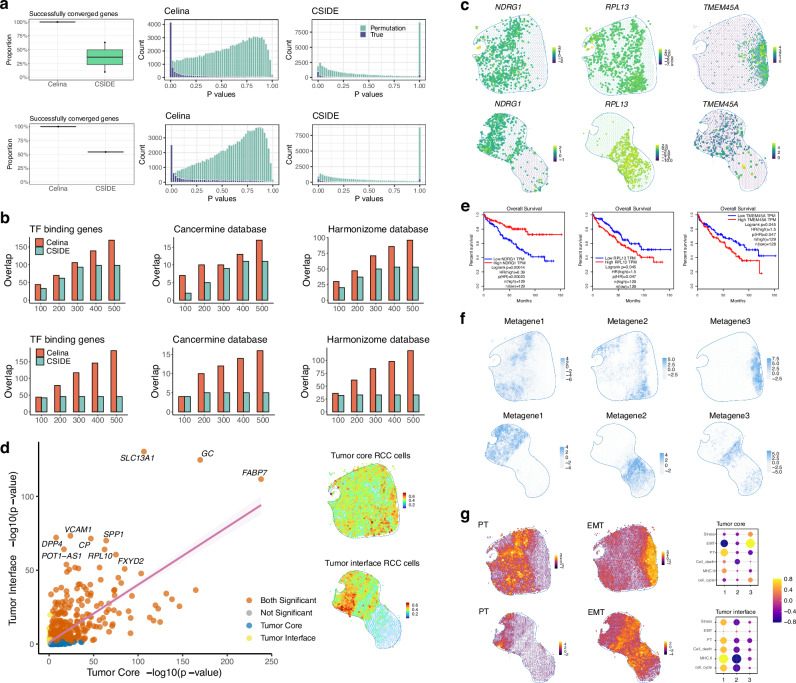
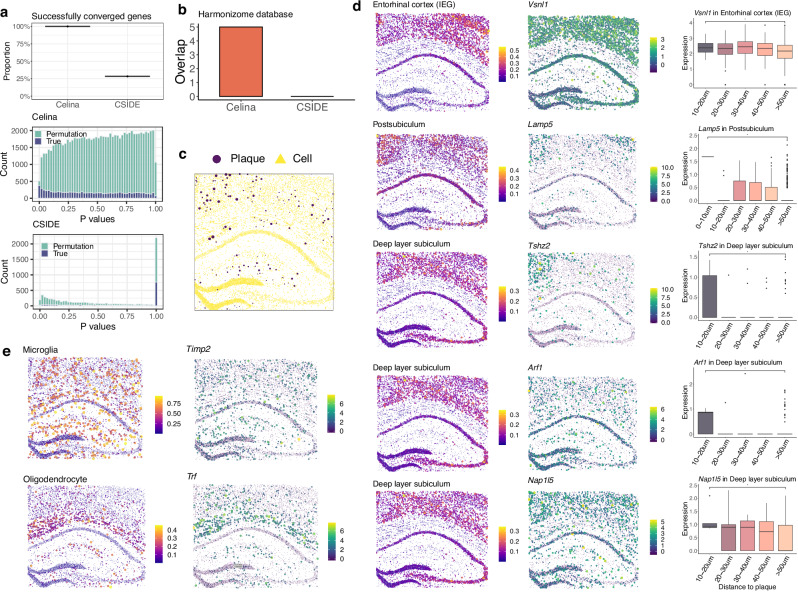
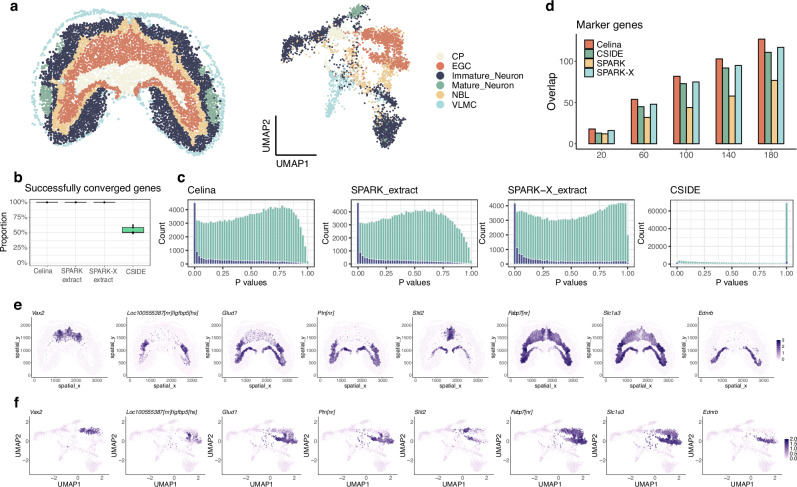
An essential task in spatial transcriptomics is identifying spatially variable genes (SVGs). Here, we present Celina, a statistical method for systematically detecting cell type-specific SVGs (ct-SVGs)-a subset of SVGs exhibiting distinct spatial expression patterns within specific cell types. Celina utilizes a spatially varying coefficient model to accurately capture each gene's spatial expression pattern in relation to the distribution of cell types across tissue locations, ensuring effective type I error control and high power. Celina proves powerful compared to existing methods in single-cell resolution spatial transcriptomics and stands as the only effective solution for spot-resolution spatial transcriptomics. Applied to five real datasets, Celina uncovers ct-SVGs associated with tumor progression and patient survival in lung cancer, identifies metagenes with unique spatial patterns linked to cell proliferation and immune response in kidney cancer, and detects genes preferentially expressed near amyloid-β plaques in an Alzheimer's model.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
GAADE: identification spatially variable genes based on adaptive graph attention network.Brief Bioinform. 2024 Nov 22;26(1):bbae669. doi: 10.1093/bib/bbae669. Brief Bioinform. 2024. PMID: 39701602 Free PMC article.
-
HEARTSVG: a fast and accurate method for identifying spatially variable genes in large-scale spatial transcriptomics.Nat Commun. 2024 Jul 7;15(1):5700. doi: 10.1038/s41467-024-49846-1. Nat Commun. 2024. PMID: 38972896 Free PMC article.
-
Evaluating spatially variable gene detection methods for spatial transcriptomics data.Genome Biol. 2024 Jan 15;25(1):18. doi: 10.1186/s13059-023-03145-y. Genome Biol. 2024. PMID: 38225676 Free PMC article.
-
Recent advances in spatially variable gene detection in spatial transcriptomics.Comput Struct Biotechnol J. 2024 Feb 2;23:883-891. doi: 10.1016/j.csbj.2024.01.016. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 38370977 Free PMC article. Review.
-
Profiling cell identity and tissue architecture with single-cell and spatial transcriptomics.Nat Rev Mol Cell Biol. 2025 Jan;26(1):11-31. doi: 10.1038/s41580-024-00768-2. Epub 2024 Aug 21. Nat Rev Mol Cell Biol. 2025. PMID: 39169166 Review.
Cited by
-
Single-cell and spatial transcriptomics profile the interaction of SPP1+ macrophages and FAP+ fibroblasts in non-small cell lung cancer.Transl Lung Cancer Res. 2025 Jul 31;14(7):2646-2669. doi: 10.21037/tlcr-2025-244. Epub 2025 Jul 25. Transl Lung Cancer Res. 2025. PMID: 40799444 Free PMC article.
-
A Complete Spatial Map of Mouse Retinal Ganglion Cells Reveals Density and Gene Expression Specializations.bioRxiv [Preprint]. 2025 Feb 12:2025.02.10.637538. doi: 10.1101/2025.02.10.637538. bioRxiv. 2025. PMID: 39990332 Free PMC article. Preprint.
-
Beyond ADME: The Endogenous Functions of Drug Transporters and Its Impact on Human Disease.Pharmaceutics. 2025 May 23;17(6):685. doi: 10.3390/pharmaceutics17060685. Pharmaceutics. 2025. PMID: 40573999 Free PMC article. Review.
-
A computational framework for estimating and testing specific parametric structures in generalized additive model.medRxiv [Preprint]. 2025 Jul 14:2025.05.12.25327450. doi: 10.1101/2025.05.12.25327450. medRxiv. 2025. PMID: 40463534 Free PMC article. Preprint.
References
-
- Wei, X. et al. Single-cell Stereo-seq reveals induced progenitor cells involved in axolotl brain regeneration. Science377, eabp9444 (2022). - PubMed
-
- Genomics, X. 10X Genomics: visium spatial gene expression. (2020).
MeSH terms
Grants and funding
- R01GM126553/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- R01HG009124/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- R01GM144960/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- R01HG011883/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- R01 HG011883/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
