

Insights into the flexibility of the domain-linking loop in actinobacterial coproheme decarboxylase through structures and molecular dynamics simulations
- PMID: 39865384
- PMCID: PMC11761711
- DOI: 10.1002/pro.70027
Insights into the flexibility of the domain-linking loop in actinobacterial coproheme decarboxylase through structures and molecular dynamics simulations
Abstract
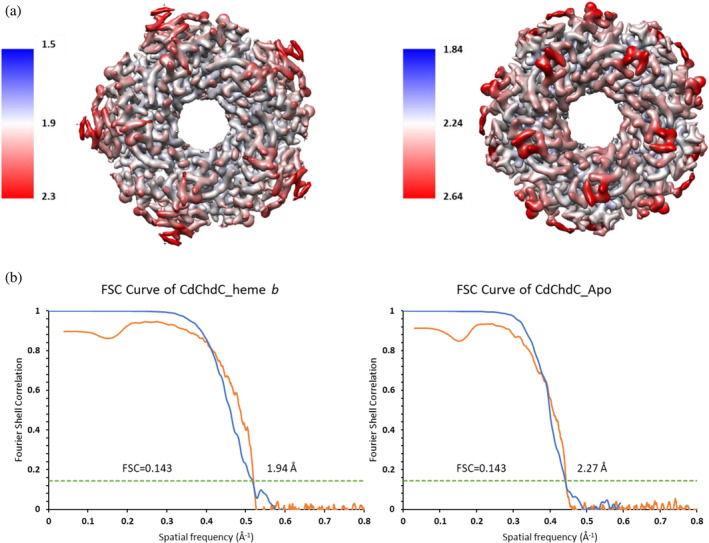
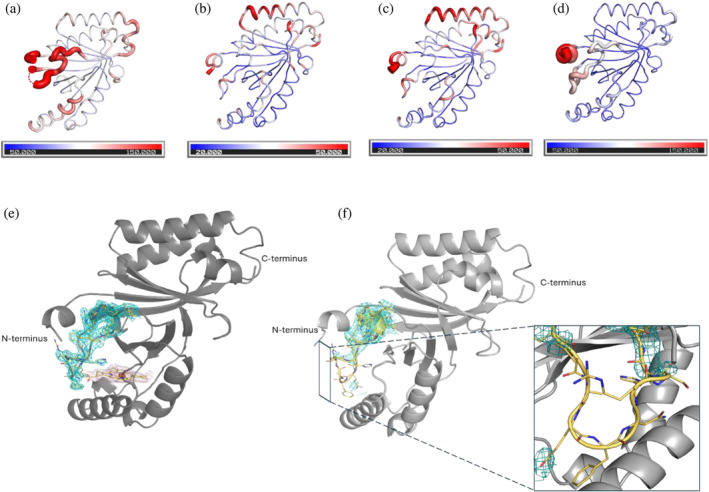
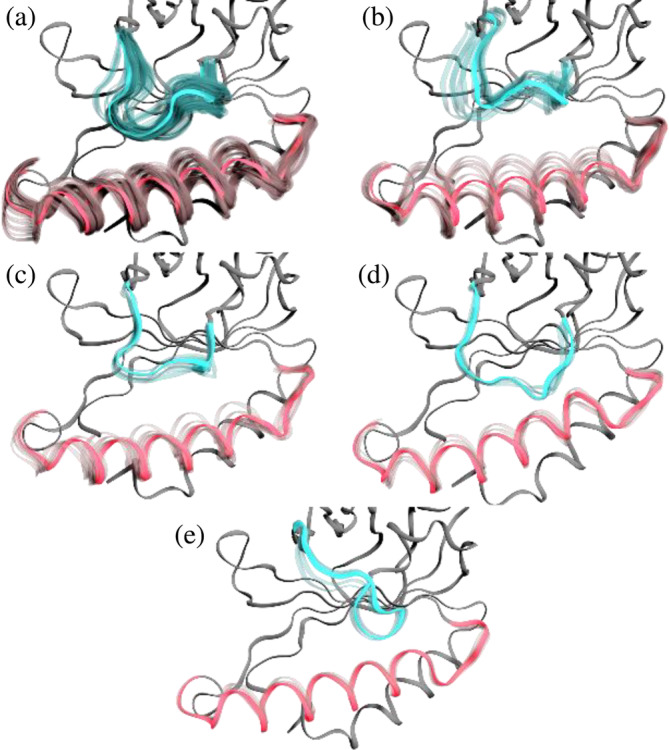
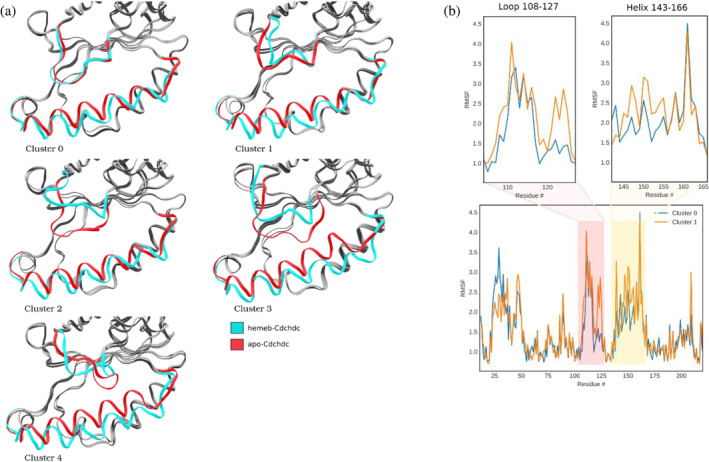
Prokaryotic heme biosynthesis in Gram-positive bacteria follows the coproporphyrin-dependent heme biosynthesis pathway. The last step in this pathway is catalyzed by the enzyme coproheme decarboxylase, which oxidatively transforms two propionate groups into vinyl groups yielding heme b. The catalytic reaction cycle of coproheme decarboxylases exhibits four different states: the apo-form, the substrate (coproheme)-bound form, a transient three-propionate intermediate form (monovinyl, monopropionate deuteroheme; MMD), and the product (heme b)-bound form. In this study, we used cryogenic electron microscopy single-particle reconstruction (cryo-EM SPR) to characterize structurally the apo and heme b-bound forms of actinobacterial coproheme decarboxylase from Corynebacterium diphtheriae. The flexible loop that connects the N-terminal and the C-terminal ferredoxin domains of coproheme decarboxylases plays an important role in interactions between the enzyme and porphyrin molecule. To understand the role of this flexible loop, we performed molecular dynamics simulations on the apo and heme b coproheme decarboxylase from Corynebacterium diphtheriae. Our results are discussed in the context of the published structural information on coproheme-bound and MMD-bound coproheme decarboxylase and with respect to the reaction mechanism. Having structural information of all four enzymatically relevant states helps in understanding structural restraints with a functional impact.
Keywords: cryo‐EM; heme biosynthesis; molecular dynamics simulations; structural biology.
© 2025 The Author(s). Protein Science published by Wiley Periodicals LLC on behalf of The Protein Society.
Conflict of interest statement
Y.G. and D.B. are co‐founders of Ligo Analytics. Y.G. serves as the CEO of Ligo Analytics.
Figures








Similar articles
-
Reactivity of Coproheme Decarboxylase with Monovinyl, Monopropionate Deuteroheme.Biomolecules. 2023 Jun 6;13(6):946. doi: 10.3390/biom13060946. Biomolecules. 2023. PMID: 37371526 Free PMC article.
-
An active site at work - the role of key residues in C. diphteriae coproheme decarboxylase.J Inorg Biochem. 2022 Apr;229:111718. doi: 10.1016/j.jinorgbio.2022.111718. Epub 2022 Jan 6. J Inorg Biochem. 2022. PMID: 35051755
-
Reaction intermediate rotation during the decarboxylation of coproheme to heme b in C. diphtheriae.Biophys J. 2021 Sep 7;120(17):3600-3614. doi: 10.1016/j.bpj.2021.06.042. Epub 2021 Jul 31. Biophys J. 2021. PMID: 34339636 Free PMC article.
-
Understanding molecular enzymology of porphyrin-binding α + β barrel proteins - One fold, multiple functions.Biochim Biophys Acta Proteins Proteom. 2021 Jan;1869(1):140536. doi: 10.1016/j.bbapap.2020.140536. Epub 2020 Sep 4. Biochim Biophys Acta Proteins Proteom. 2021. PMID: 32891739 Free PMC article. Review.
-
HemQ: An iron-coproporphyrin oxidative decarboxylase for protoheme synthesis in Firmicutes and Actinobacteria.Arch Biochem Biophys. 2015 May 15;574:27-35. doi: 10.1016/j.abb.2015.02.017. Epub 2015 Feb 21. Arch Biochem Biophys. 2015. PMID: 25711532 Free PMC article. Review.
References
-
- Berendsen HJC, Postma JPM, Van Gunsteren WF, Dinola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690.
-
- Brooks CL. Computer simulation of liquids. J Solution Chem. 1989;18:99.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
