

Red-shifted optical absorption induced by donor-acceptor-donor π-extended dibenzalacetone derivatives
- PMID: 39867319
- PMCID: PMC11758225
- DOI: 10.1039/d4ra07256a
Red-shifted optical absorption induced by donor-acceptor-donor π-extended dibenzalacetone derivatives
Abstract
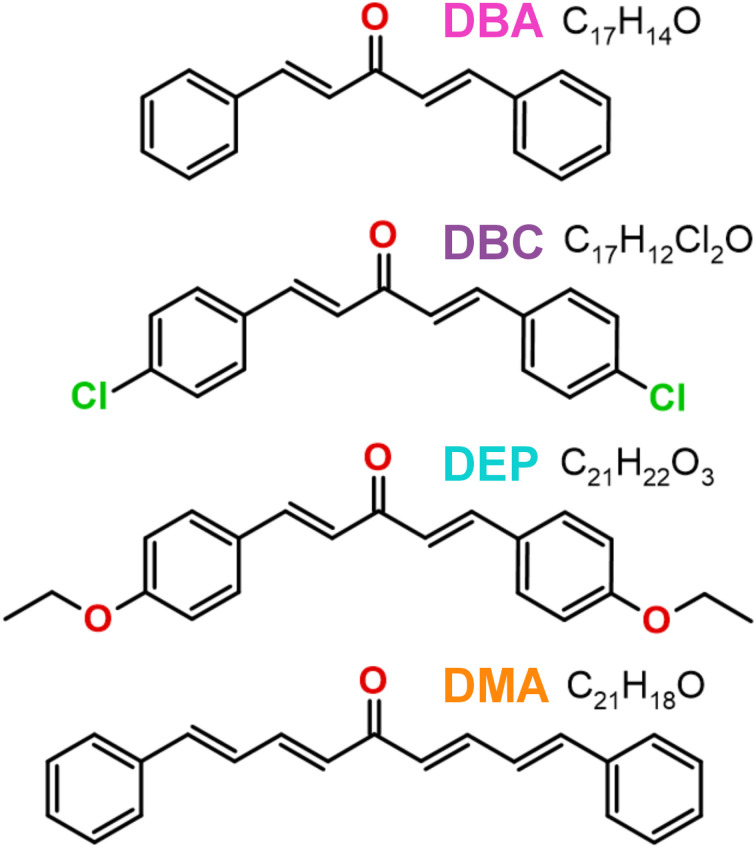
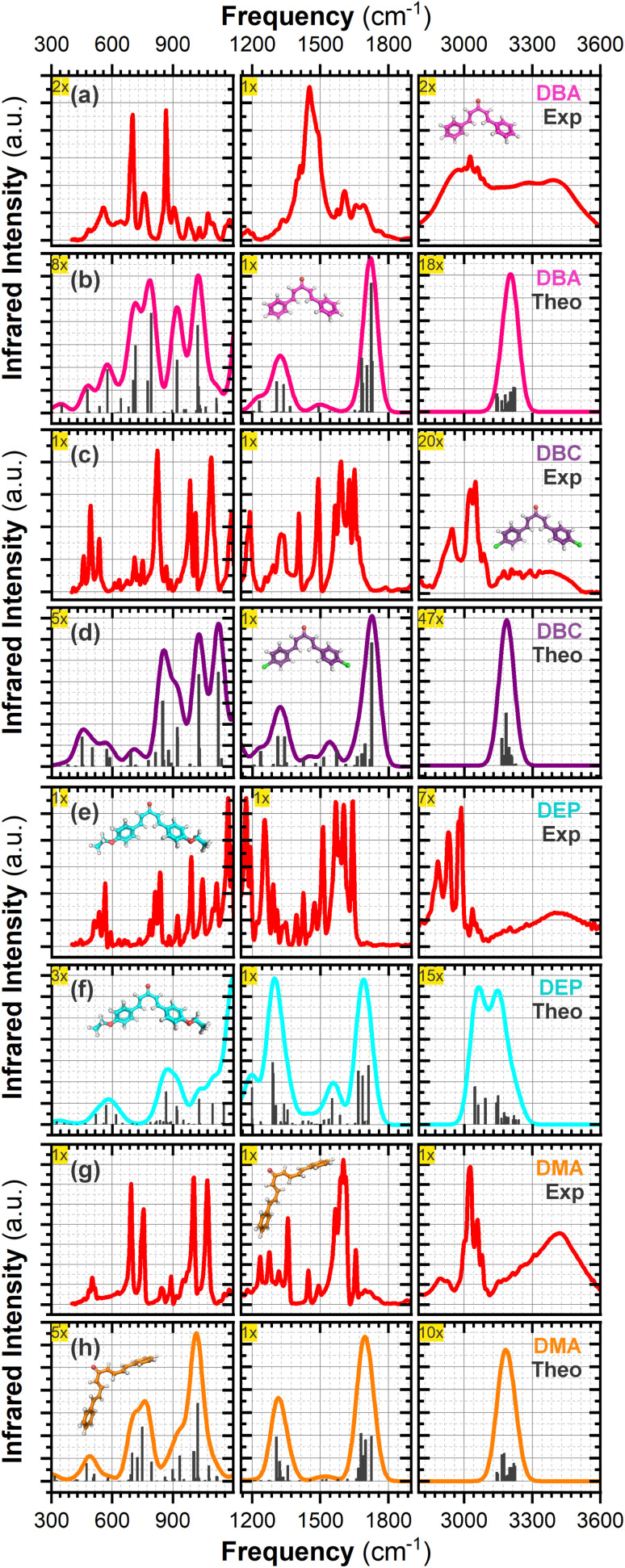
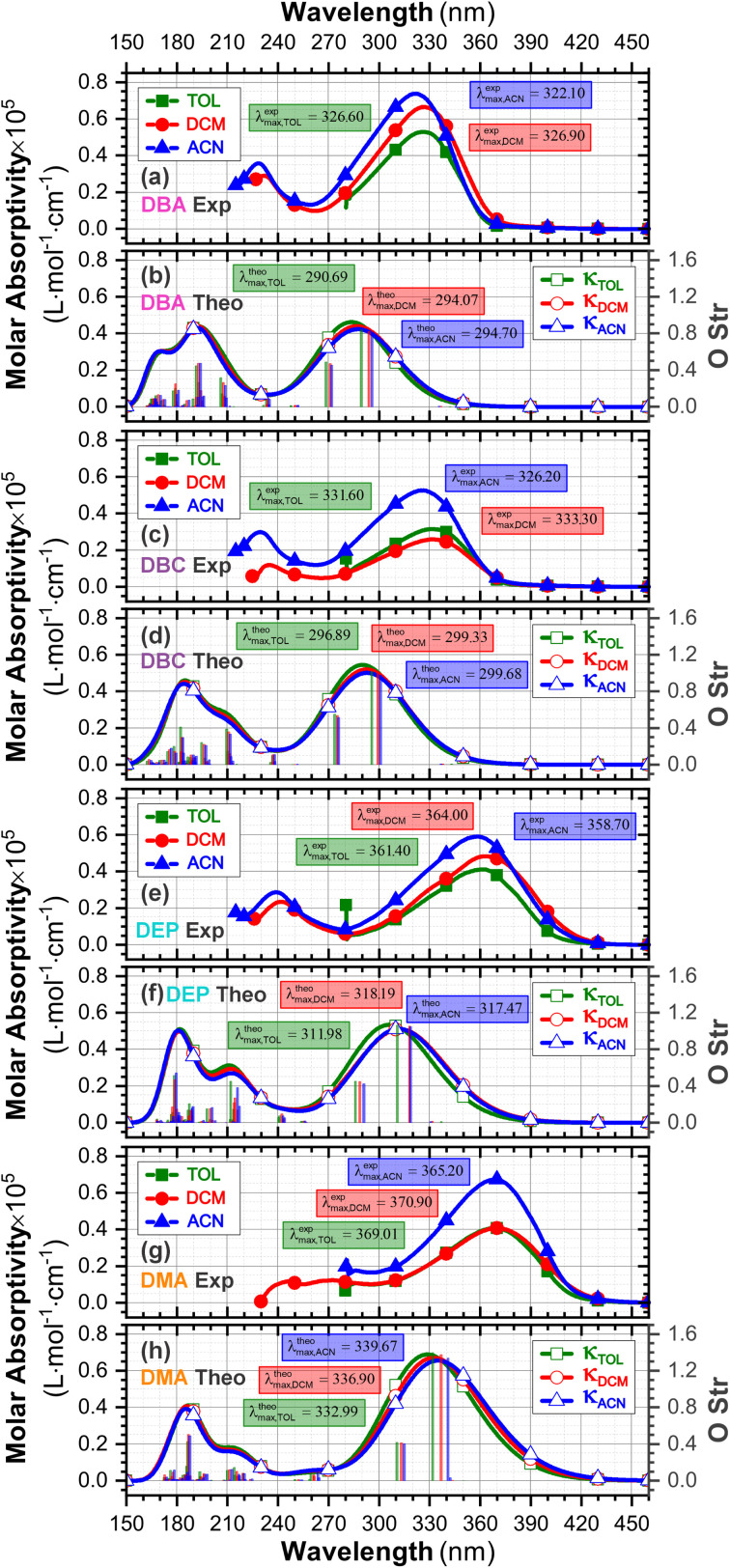
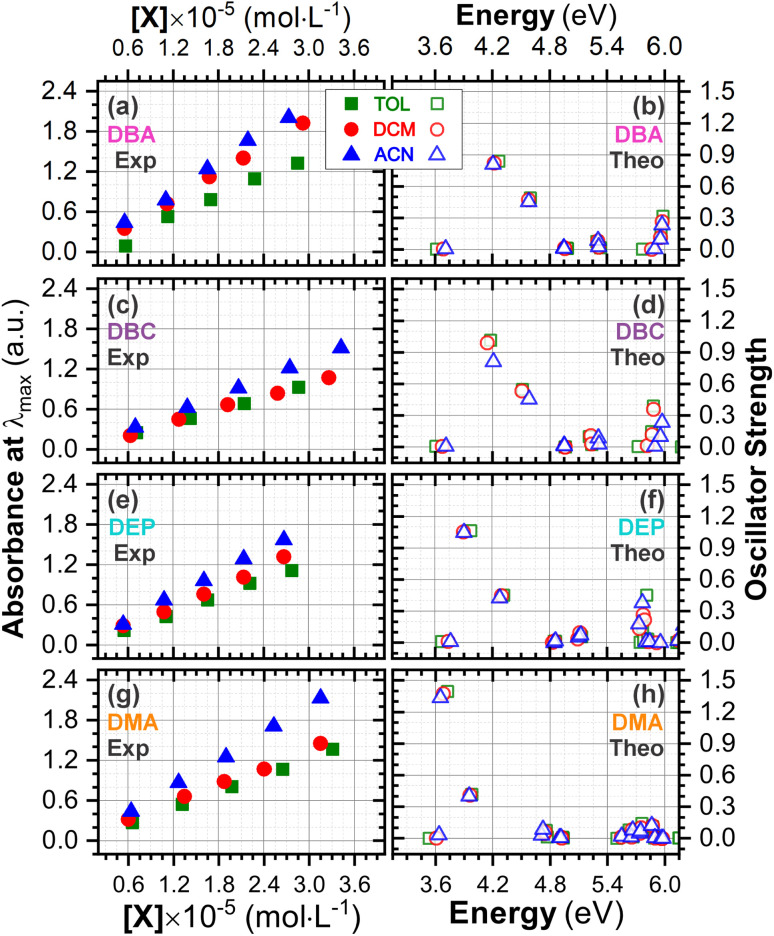
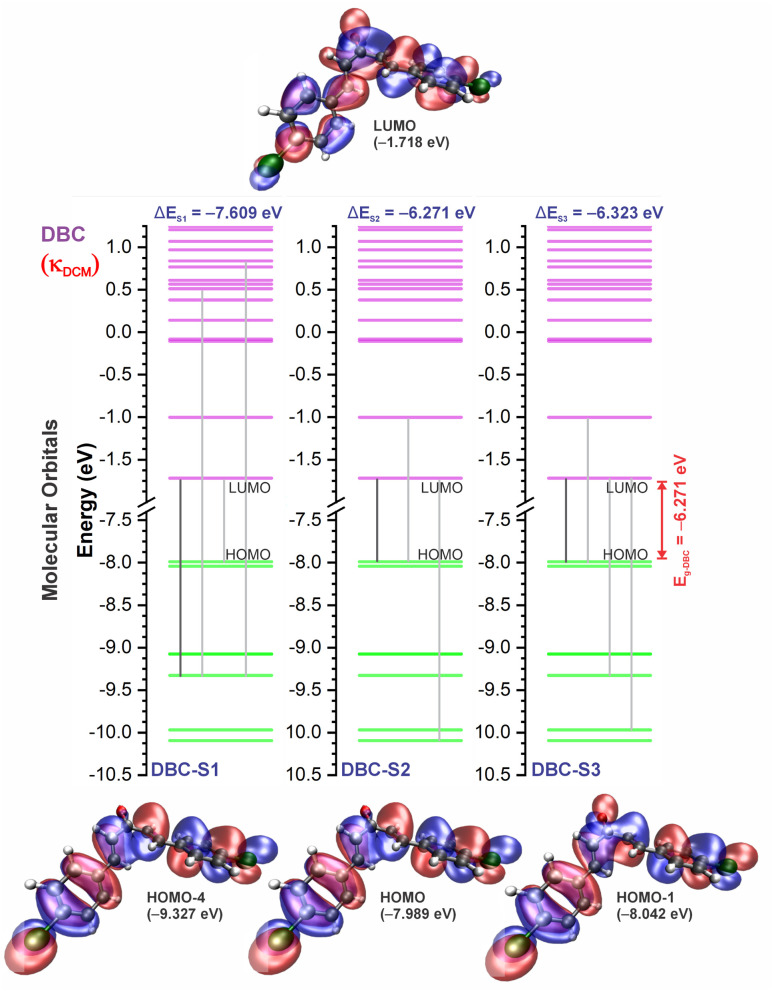
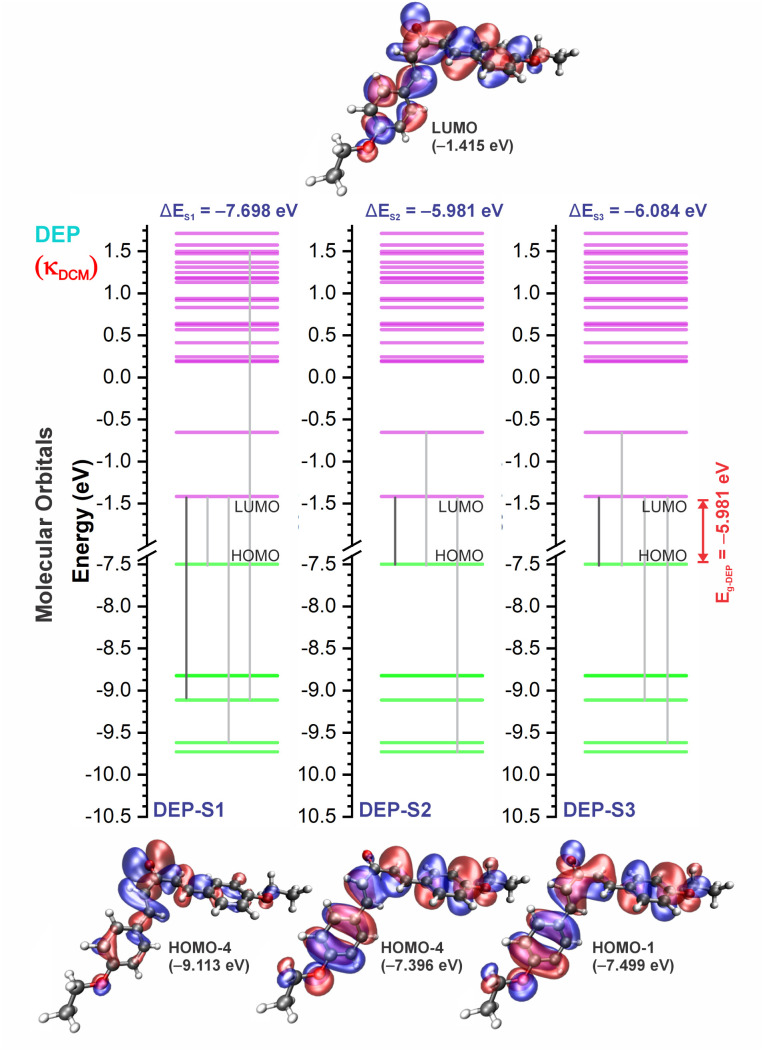
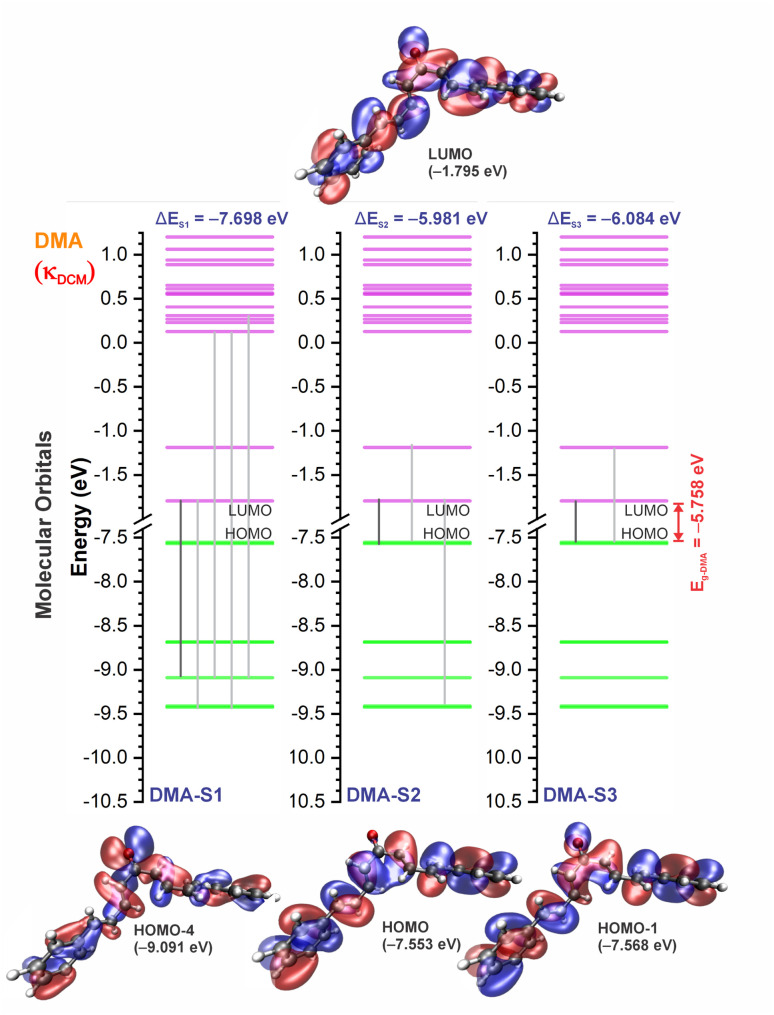
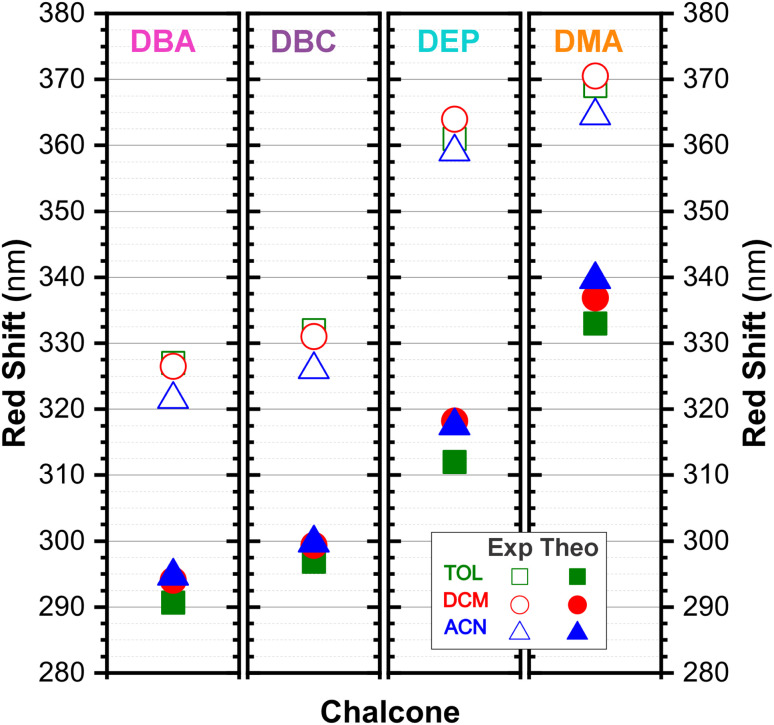
Chalcones demonstrate significant absorption in the near ultraviolet-visible spectrum, making them valuable for applications such as solar cells, light-emitting diodes, and nonlinear optics. This study investigates four dibenzalacetone derivatives (DBAd), DBA, DBC, DEP, and DMA, examining the impact of electron-donating and electron-withdrawing groups and conjugation elongation on their electronic structure in solvents of varying polarities. Using the Polarizable Continuum Model (PCM) and time-dependent density functional theory (TD-DFT), we characterized the excited states of these compounds. Our results reveal a consistent red-shift in the absorption spectrum, with electron-donating groups like ethoxy inducing a more pronounced red-shift than chlorine. Extending conjugation in DMA further shifted the absorption band to lower energy. Solvatochromism influenced the absorption intensities, underscoring the importance of evaluating parameters beyond λ max. Although our methodologies provided a satisfactory correlation between theoretical and experimental data, they also indicate the need for further theoretical models to accurately capture solute-solvent interactions and describe charge-separated states. The results indicated that dibenzalacetone derivatives have potential as alternative materials for development of organic solar cells.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures


Similar articles
-
DFT and TD-DFT Investigations for the Limitations of Lengthening the Polyene Bridge between N,N-dimethylanilino Donor and Dicyanovinyl Acceptor Molecules as a D-π-A Dye-Sensitized Solar Cell.Int J Mol Sci. 2024 May 21;25(11):5586. doi: 10.3390/ijms25115586. Int J Mol Sci. 2024. PMID: 38891775 Free PMC article.
-
Experimental and theoretical study of donor-π-acceptor compounds based on malononitrile.Chem Cent J. 2018 Mar 9;12(1):26. doi: 10.1186/s13065-018-0394-5. Chem Cent J. 2018. PMID: 29524022 Free PMC article.
-
DFT and TD-DFT calculation of new thienopyrazine-based small molecules for organic solar cells.Chem Cent J. 2016 Oct 27;10:67. doi: 10.1186/s13065-016-0216-6. eCollection 2016. Chem Cent J. 2016. PMID: 27843488 Free PMC article.
-
Donor-acceptor substituted phenylethynyltriphenylenes - excited state intramolecular charge transfer, solvatochromic absorption and fluorescence emission.Beilstein J Org Chem. 2010 Oct 18;6:992-1001. doi: 10.3762/bjoc.6.112. Beilstein J Org Chem. 2010. PMID: 21085512 Free PMC article.
-
Molecular design of D-π-A-π-D small molecule donor materials with narrow energy gap for organic solar cells applications.J Mol Model. 2023 Aug 5;29(9):273. doi: 10.1007/s00894-023-05680-8. J Mol Model. 2023. PMID: 37542668
References
-
- Ostroverkhova O. Chem. Rev. 2016;116:13279–13412. - PubMed
-
- Kukhta N. A. Bryce M. R. Mater. Horiz. 2021;8:33–55. - PubMed
-
- Hirono A. Sakai H. Kochi S. Sato T. Hasobe T. J. Phys. Chem. B. 2020;124:9921–9930. - PubMed
-
- Mellado M. Sariego-Kluge R. Valdés-Navarro F. González C. Sánchez-González R. Pizarro N. Villena J. Jara-Gutierrez C. Cordova C. Bravo M. A. et al. . Spectrochim. Acta, Part A. 2023;291:122332. - PubMed
-
- Shafiq I. Khalid M. Muneer M. Asghar M. A. Baby R. Ahmed S. Ahamad T. de Alcântara Morais S. F. Braga A. A. Mater. Chem. Phys. 2023;308:128154.
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous