

IFN-γ licenses normal and pathogenic ALPK1/TIFA pathway in human monocytes
- PMID: 39868044
- PMCID: PMC11758396
- DOI: 10.1016/j.isci.2024.111563
IFN-γ licenses normal and pathogenic ALPK1/TIFA pathway in human monocytes
Abstract
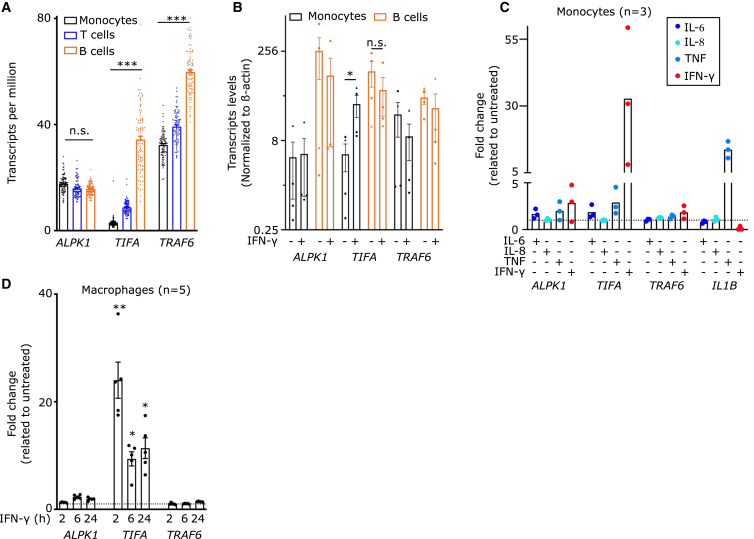
Alpha-kinase 1 (ALPK1) is an immune receptor sensing the bacterial nucleotide sugar ADP-heptose. ALPK1 phosphorylates TIFA leading to its oligomerization and downstream NF-κB activation. Specific mutations in ALPK1 are associated with an autoinflammatory syndrome termed ROSAH and with spiradenoma (skin cancers with sweat gland differentiation). This study investigated ALPK1 responses in human mononuclear cells and demonstrates that human mononuclear cells have distinct abilities to respond to ADP-heptose. Notably, IFN-γ is required to license the ALPK1/TIFA pathway in monocytes, while it was dispensable for the responsiveness of B cells. IFN-γ induced TIFA upregulation in monocytes, and TIFA induction was sufficient to recapitulate the licensing effect of IFN-γ. IFN-γ treatment promoted the phenotypic expression of pathogenic ALPK1 mutations. The licensing effect of IFN-γ in monocytes was blocked by JAK inhibitors. These findings underscore the critical role of IFN-γ in ALPK1 function and suggest JAK inhibitors as potential therapies for ALPK1-related inflammatory conditions.
Keywords: Biological sciences; Immunology; Natural sciences.
© 2024 The Author(s).
Conflict of interest statement
T.H. team obtained a research contract with financial support from Drug Farm, a company that develops agonists and inhibitors of ALPK1. Y.J. received consultant fees from Drug Farm. Other authors did not report any conflict of interest related to the present work.
Figures





References
-
- Mattioli I., Sebald A., Bucher C., Charles R.-P., Nakano H., Doi T., Kracht M., Schmitz M.L. Transient and Selective NF-κB p65 Serine 536 Phosphorylation Induced by T Cell Costimulation Is Mediated by IκB Kinase β and Controls the Kinetics of p65 Nuclear Import 1. J. Immunol. 2004;172:6336–6344. doi: 10.4049/jimmunol.172.10.6336. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
