

This is a preprint.
Defining the Polycystin Pharmacophore Through HTS & Computational Biophysics
- PMID: 39868095
- PMCID: PMC11761769
- DOI: 10.1101/2025.01.13.632808
Defining the Polycystin Pharmacophore Through HTS & Computational Biophysics
Update in
-
Defining the polycystin pharmacophore through high-throughput screening and computational biophysics.Br J Pharmacol. 2025 Oct;182(19):4611-4624. doi: 10.1111/bph.70080. Epub 2025 Jun 15. Br J Pharmacol. 2025. PMID: 40518131
Abstract
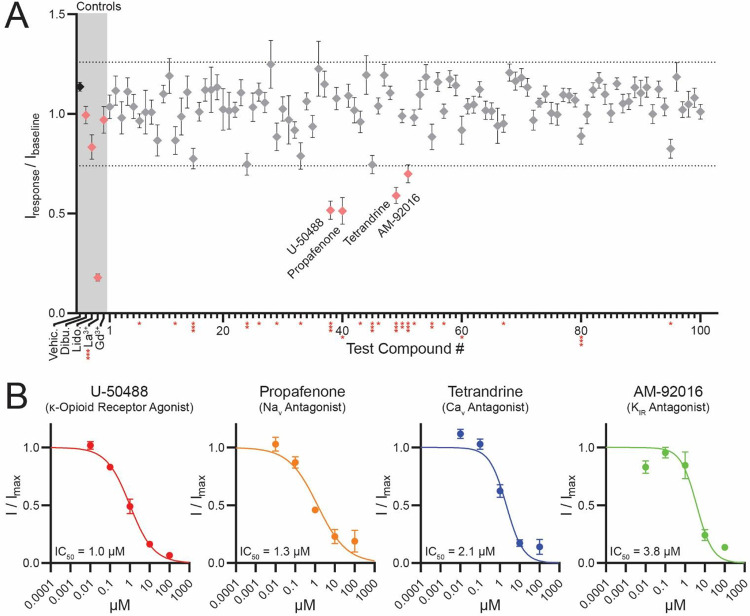
Background and purpose: Polycystins (PKD2, PKD2L1) are voltage-gated and Ca2+-modulated members of the transient receptor potential (TRP) family of ion channels. Loss of PKD2L1 expression results in seizure-susceptibility and autism-like features in mice, whereas variants in PKD2 cause autosomal dominant polycystic kidney disease. Despite decades of evidence clearly linking their dysfunction to human disease and demonstrating their physiological importance in the brain and kidneys, the polycystin pharmacophore remains undefined. Contributing to this knowledge gap is their resistance to drug screening campaigns, which are hindered by these channels' unique subcellular trafficking to organelles such as the primary cilium. PKD2L1 is the only member of the polycystin family to form constitutively active ion channels on the plasma membrane when overexpressed.
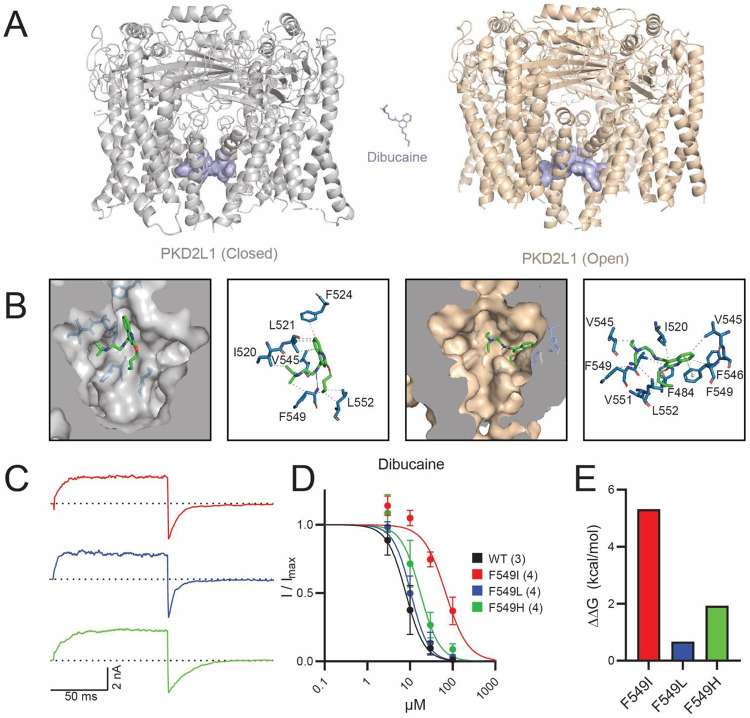
Experimental approach: HEK293 cells stably expressing PKD2L1 F514A were pharmacologically screened via high-throughput electrophysiology to identify potent polycystin channel modulators. In-silico docking analysis and mutagenesis were used to define the receptor sites of screen hits. Inhibition by membrane-impermeable QX-314 was used to evaluate PKD2L1's binding site accessibility.
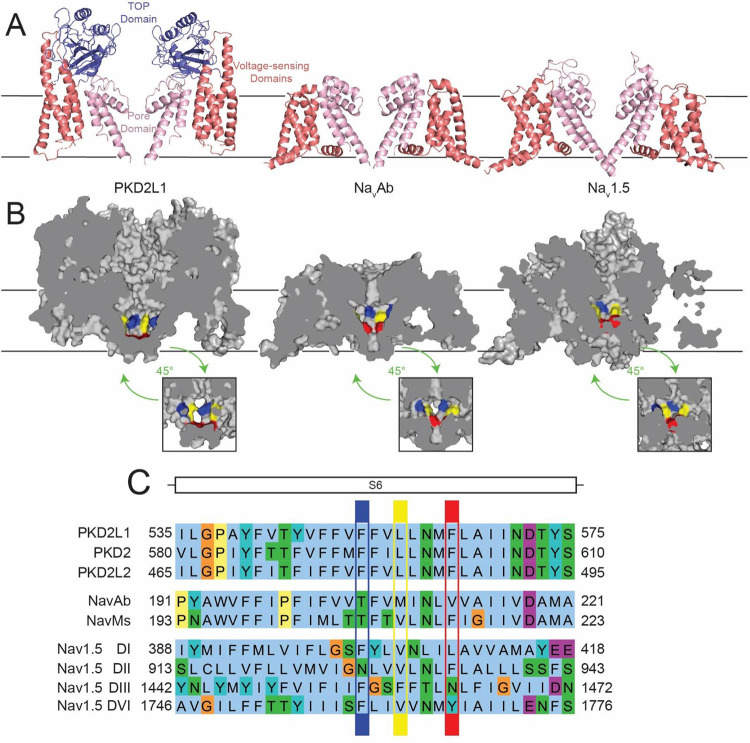
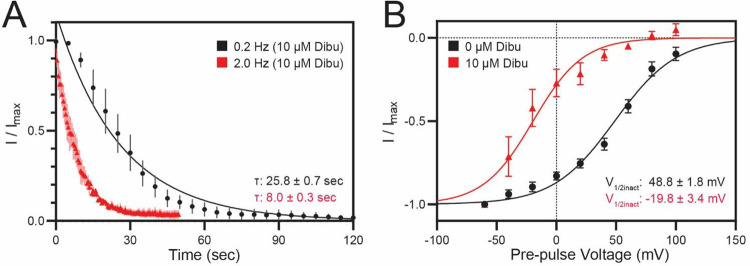
Key results: Screen results identify potent PKD2L1 antagonists with divergent chemical core structures and highlight striking similarities between the molecular pharmacology of PKD2L1 and voltage-gated sodium channels. Docking analysis, channel mutagenesis, and physiological recordings identify an open-state accessible lateral fenestration receptor within the pore, and a mechanism of inhibition that stabilizes the PKD2L1 inactivated state.
Conclusion and implication: Outcomes establish the suitability of our approach to expand our chemical knowledge of polycystins and delineates novel receptor moieties for the development of channel-specific antagonists in TRP channel research.
Keywords: ADPKD; Computational Biophysics; High-Throughput Electrophysiology; Molecular Docking; PKD2; PKD2L1; Pharmacology; TRP channels; autosomal polycystic kidney disease; ion channels; molecular mechanisms; polycystin; primary cilia.
Conflict of interest statement
Conflict of Interest Statement: The authors declare that they have no conflict of interest
Figures


References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous