

methylGrapher: genome-graph-based processing of DNA methylation data from whole genome bisulfite sequencing
- PMID: 39868538
- PMCID: PMC11770346
- DOI: 10.1093/nar/gkaf028
methylGrapher: genome-graph-based processing of DNA methylation data from whole genome bisulfite sequencing
Abstract
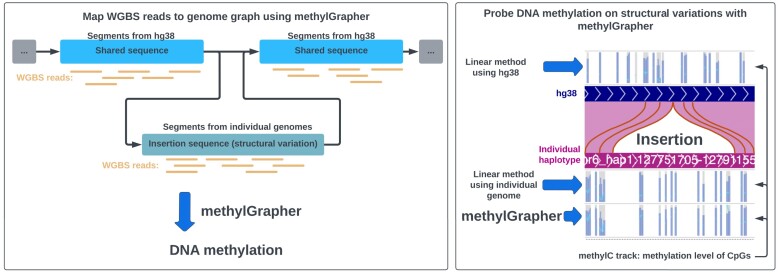
Genome graphs, including the recently released draft human pangenome graph, can represent the breadth of genetic diversity and thus transcend the limits of traditional linear reference genomes. However, there are no genome-graph-compatible tools for analyzing whole genome bisulfite sequencing (WGBS) data. To close this gap, we introduce methylGrapher, a tool tailored for accurate DNA methylation analysis by mapping WGBS data to a genome graph. Notably, methylGrapher can reconstruct methylation patterns along haplotype paths precisely and efficiently. To demonstrate the utility of methylGrapher, we analyzed the WGBS data derived from five individuals whose genomes were included in the first Human Pangenome draft as well as WGBS data from ENCODE (EN-TEx). Along with standard performance benchmarking, we show that methylGrapher fully recapitulates DNA methylation patterns defined by classic linear genome analysis approaches. Importantly, methylGrapher captures a substantial number of CpG sites that are missed by linear methods, and improves overall genome coverage while reducing alignment reference bias. Thus, methylGrapher is a first step toward unlocking the full potential of Human Pangenome graphs in genomic DNA methylation analysis.
© The Author(s) 2025. Published by Oxford University Press on behalf of Nucleic Acids Research.
Conflict of interest statement
None declared.
Figures






References
-
- Lander ES, Linton LM, Birren B et al. . Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
