

Gastric cancer genomics study using reference human pangenomes
- PMID: 39870503
- PMCID: PMC11772497
- DOI: 10.26508/lsa.202402977
Gastric cancer genomics study using reference human pangenomes
Abstract
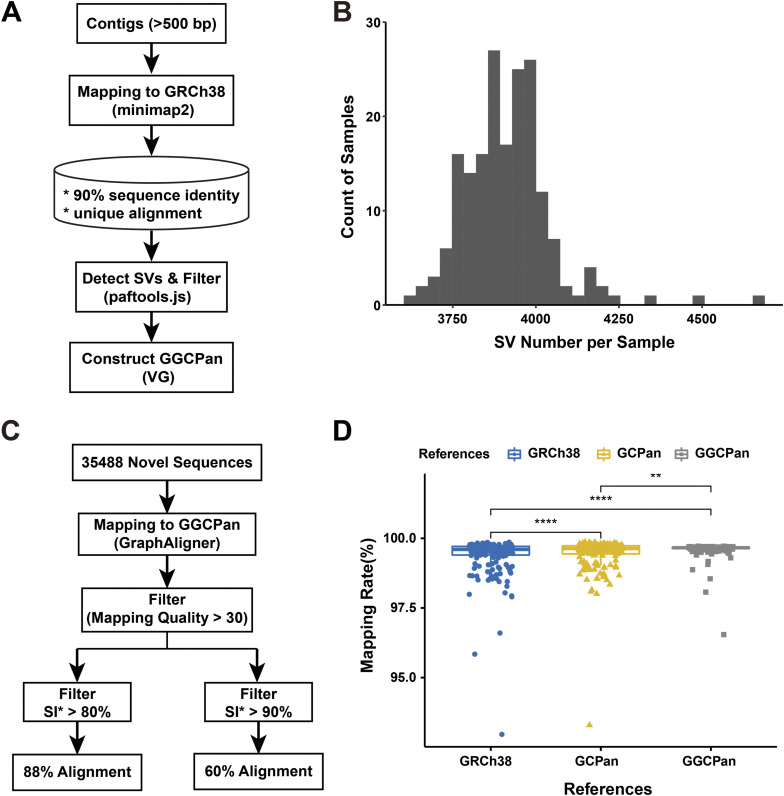
A pangenome is the sum of the genetic information of all individuals in a species or a population. Genomics research has been gradually shifted to a paradigm using a pangenome as the reference. However, in disease genomics study, pangenome-based analysis is still in its infancy. In this study, we introduced a graph-based pangenome GGCPan from 185 patients with gastric cancer. We then systematically compared the cancer genomics study results using GGCPan, a linear pangenome GCPan, and the human reference genome as the reference. For small variant detection and microsatellite instability status identification, there is little difference in using three different genomes. Using GGCPan as the reference had a significant advantage in structural variant identification. A total of 24 candidate gastric cancer driver genes were detected using three different reference genomes, of which eight were common and five were detected only based on pangenomes. Our results showed that disease-specific pangenome as a reference is promising and a whole set of tools are still to be developed or improved for disease genomics study in the pangenome era.
© 2025 Jiao et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures










Similar articles
-
Pangenome graphs in infectious disease: a comprehensive genetic variation analysis of Neisseria meningitidis leveraging Oxford Nanopore long reads.Front Genet. 2023 Aug 10;14:1225248. doi: 10.3389/fgene.2023.1225248. eCollection 2023. Front Genet. 2023. PMID: 37636268 Free PMC article.
-
A pangenome analysis pipeline provides insights into functional gene identification in rice.Genome Biol. 2023 Jan 26;24(1):19. doi: 10.1186/s13059-023-02861-9. Genome Biol. 2023. PMID: 36703158 Free PMC article.
-
What Are We Learning from Plant Pangenomes?Annu Rev Plant Biol. 2025 May;76(1):663-686. doi: 10.1146/annurev-arplant-090823-015358. Epub 2024 Dec 2. Annu Rev Plant Biol. 2025. PMID: 39621536 Review.
-
A stepwise guide for pangenome development in crop plants: an alfalfa (Medicago sativa) case study.BMC Genomics. 2024 Oct 31;25(1):1022. doi: 10.1186/s12864-024-10931-w. BMC Genomics. 2024. PMID: 39482604 Free PMC article. Review.
-
Perspectives and opportunities in forensic human, animal, and plant integrative genomics in the Pangenome era.Forensic Sci Int. 2025 Feb;367:112370. doi: 10.1016/j.forsciint.2025.112370. Epub 2025 Jan 12. Forensic Sci Int. 2025. PMID: 39813779 Review.
References
-
- Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, et al. (1998) A national cancer institute workshop on microsatellite instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58: 5248–5257. - PubMed
-
- Bonneville R, Krook MA, Chen H-Z, Smith A, Samorodnitsky E, Wing MR, Reeser JW, Roychowdhury S (2020) Detection of microsatellite instability biomarkers via next-generation sequencing. In Biomarkers for Immunotherapy of Cancer: Methods and Protocols, Thurin M, Cesano A, Marincola FM (eds), Vol 2055, pp 119–132. New York, NY: Springer. - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical