

A genome-wide atlas of human cell morphology
- PMID: 39870862
- PMCID: PMC11903339
- DOI: 10.1038/s41592-024-02537-7
A genome-wide atlas of human cell morphology
Abstract
A key challenge of the modern genomics era is developing empirical data-driven representations of gene function. Here we present the first unbiased morphology-based genome-wide perturbation atlas in human cells, containing three genome-wide genotype-phenotype maps comprising CRISPR-Cas9-based knockouts of >20,000 genes in >30 million cells. Our optical pooled cell profiling platform (PERISCOPE) combines a destainable high-dimensional phenotyping panel (based on Cell Painting) with optical sequencing of molecular barcodes and a scalable open-source analysis pipeline to facilitate massively parallel screening of pooled perturbation libraries. This perturbation atlas comprises high-dimensional phenotypic profiles of individual cells with sufficient resolution to cluster thousands of human genes, reconstruct known pathways and protein-protein interaction networks, interrogate subcellular processes and identify culture media-specific responses. Using this atlas, we identify the poorly characterized disease-associated TMEM251/LYSET as a Golgi-resident transmembrane protein essential for mannose-6-phosphate-dependent trafficking of lysosomal enzymes. In sum, this perturbation atlas and screening platform represents a rich and accessible resource for connecting genes to cellular functions at scale.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: C.H.J. and J.Y. are employees of Calico Life Sciences LLC. S.S. and A.E.C. serve as scientific advisors for companies that use image-based profiling and Cell Painting (A.E.C: Recursion, SyzOnc and S.S.: Waypoint Bio, Dewpoint Therapeutics) and receive honoraria for occasional talks at pharmaceutical and biotechnology companies. P.C.B. is a consultant to or holds equity in 10X Genomics, General Automation Lab Technologies/Isolation Bio, Celsius Therapeutics, Next Gen Diagnostics, Cache DNA, Concerto Biosciences, Stately, Ramona Optics, Bifrost Biosystems and Amber Bio. The laboratory of P.C.B. also received research funding from Merck and Genentech for work related to genetic screening. The Broad Institute and MIT may seek to commercialize aspects of this work, and related applications for intellectual property have been filed, including WO2019222284A1 ‘In situ cell screening methods and systems’. The other authors declare no competing interests.
Figures
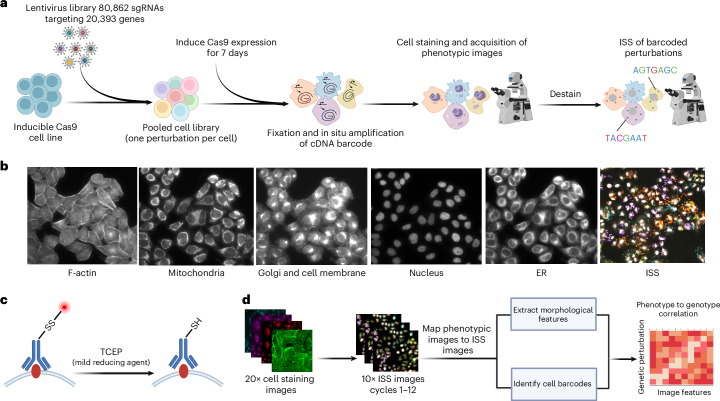
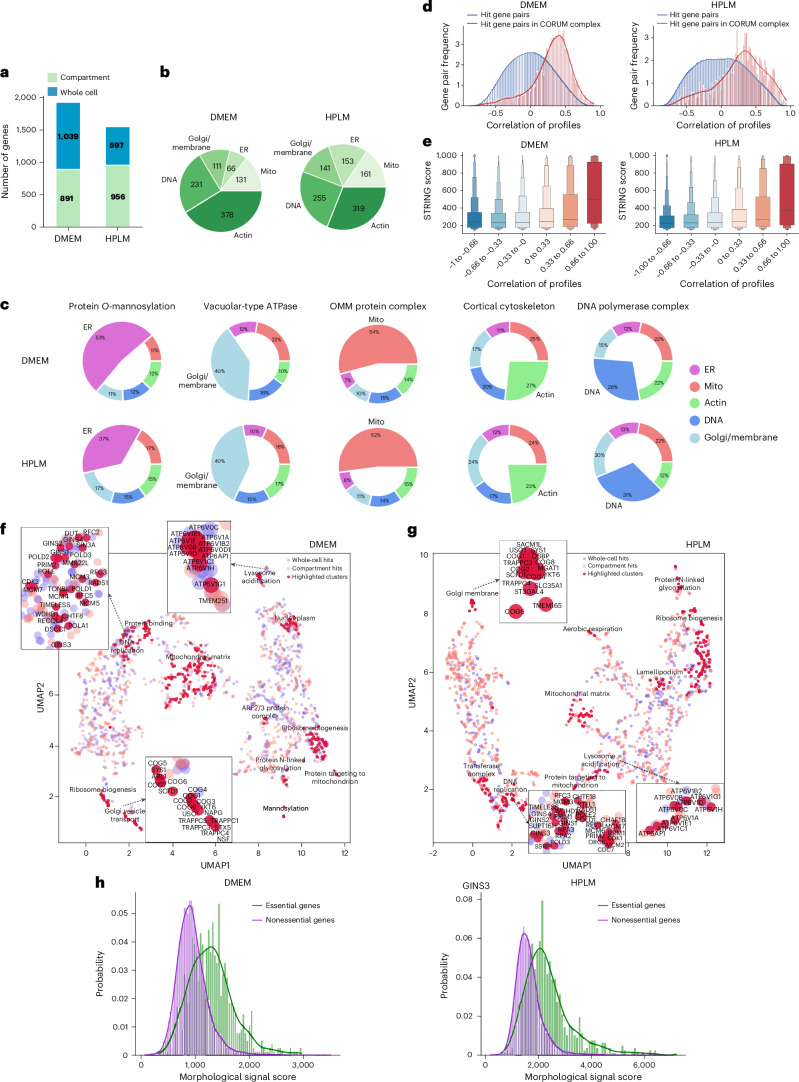


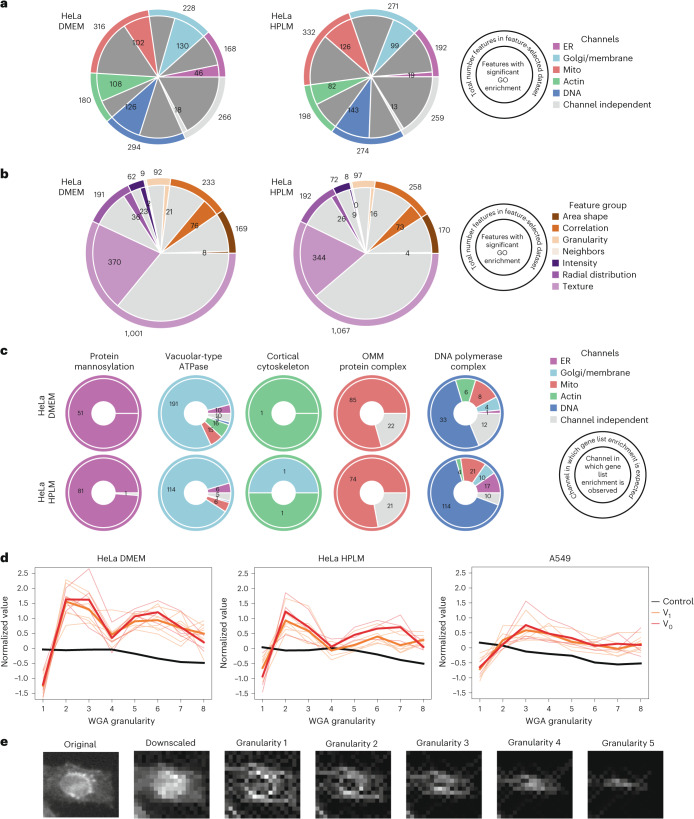








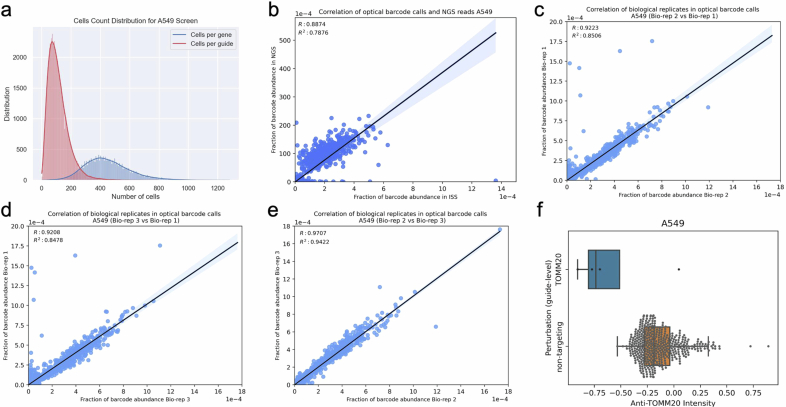


Update of
-
A genome-wide atlas of human cell morphology.bioRxiv [Preprint]. 2023 Aug 7:2023.08.06.552164. doi: 10.1101/2023.08.06.552164. bioRxiv. 2023. Update in: Nat Methods. 2025 Mar;22(3):621-633. doi: 10.1038/s41592-024-02537-7. PMID: 37609130 Free PMC article. Updated. Preprint.
References
-
- Doench, J. G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet.19, 67–80 (2018). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
