

Multiplexed spatial mapping of chromatin features, transcriptome and proteins in tissues
- PMID: 39870864
- PMCID: PMC11906265
- DOI: 10.1038/s41592-024-02576-0
Multiplexed spatial mapping of chromatin features, transcriptome and proteins in tissues
Abstract
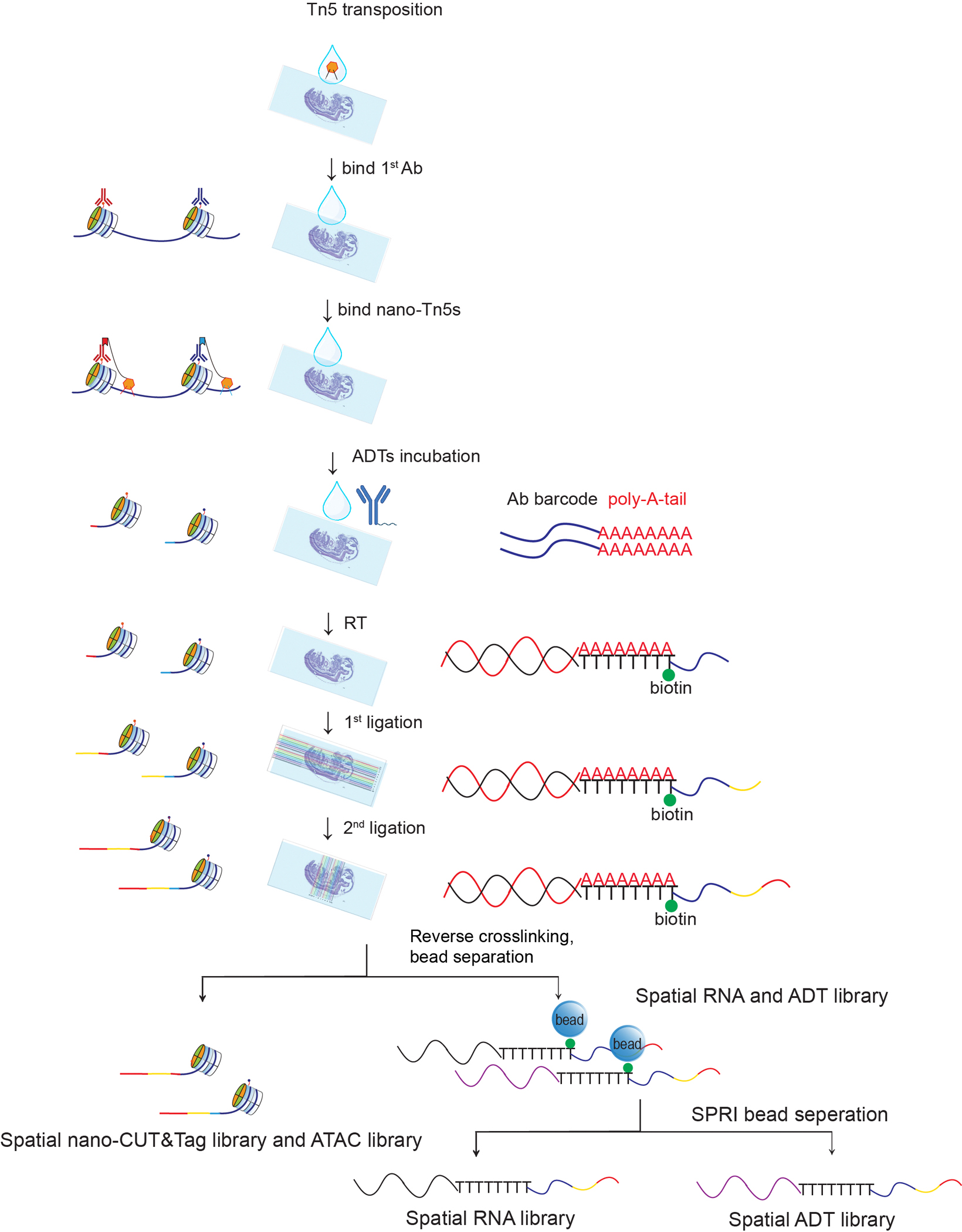
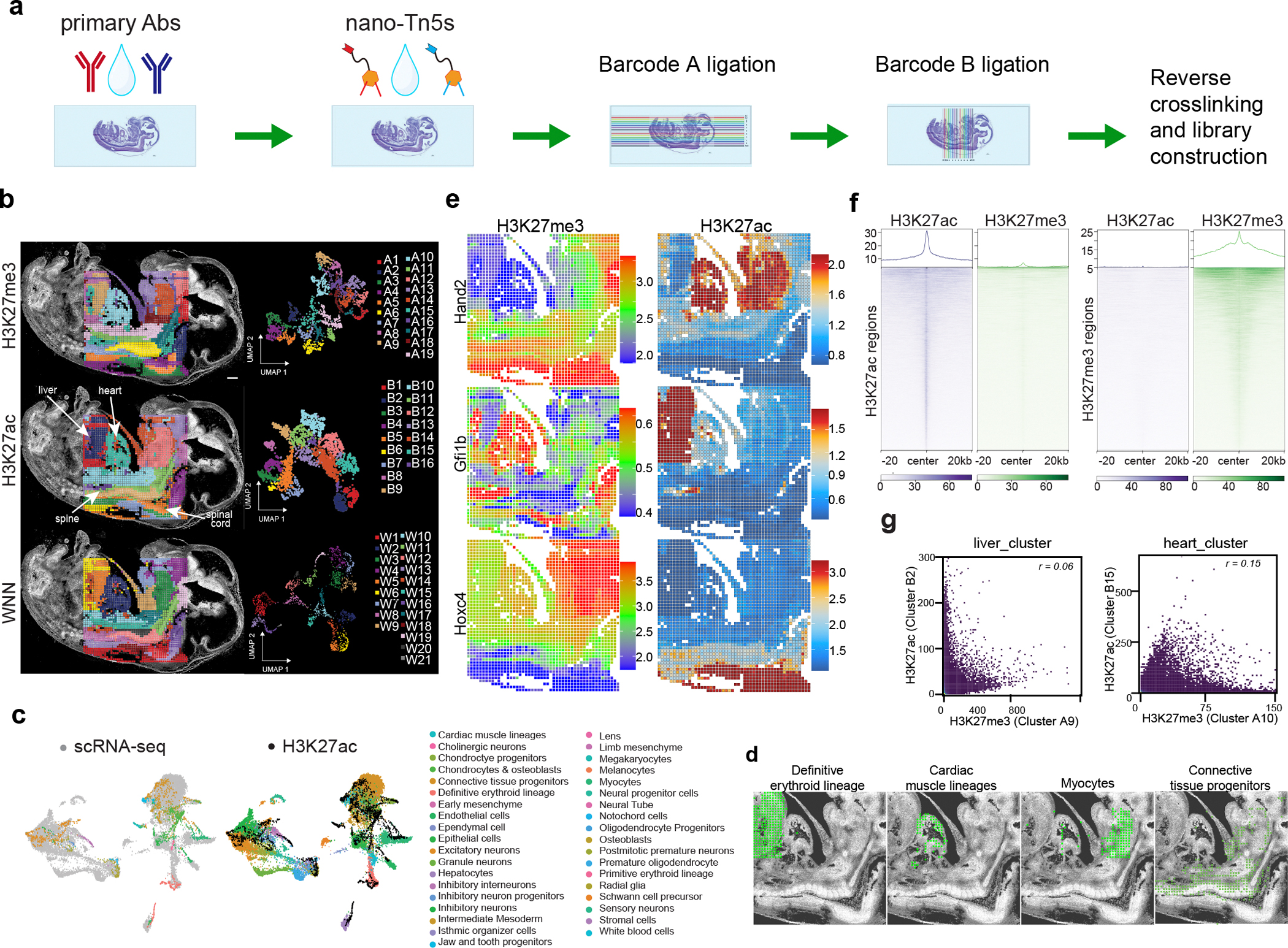
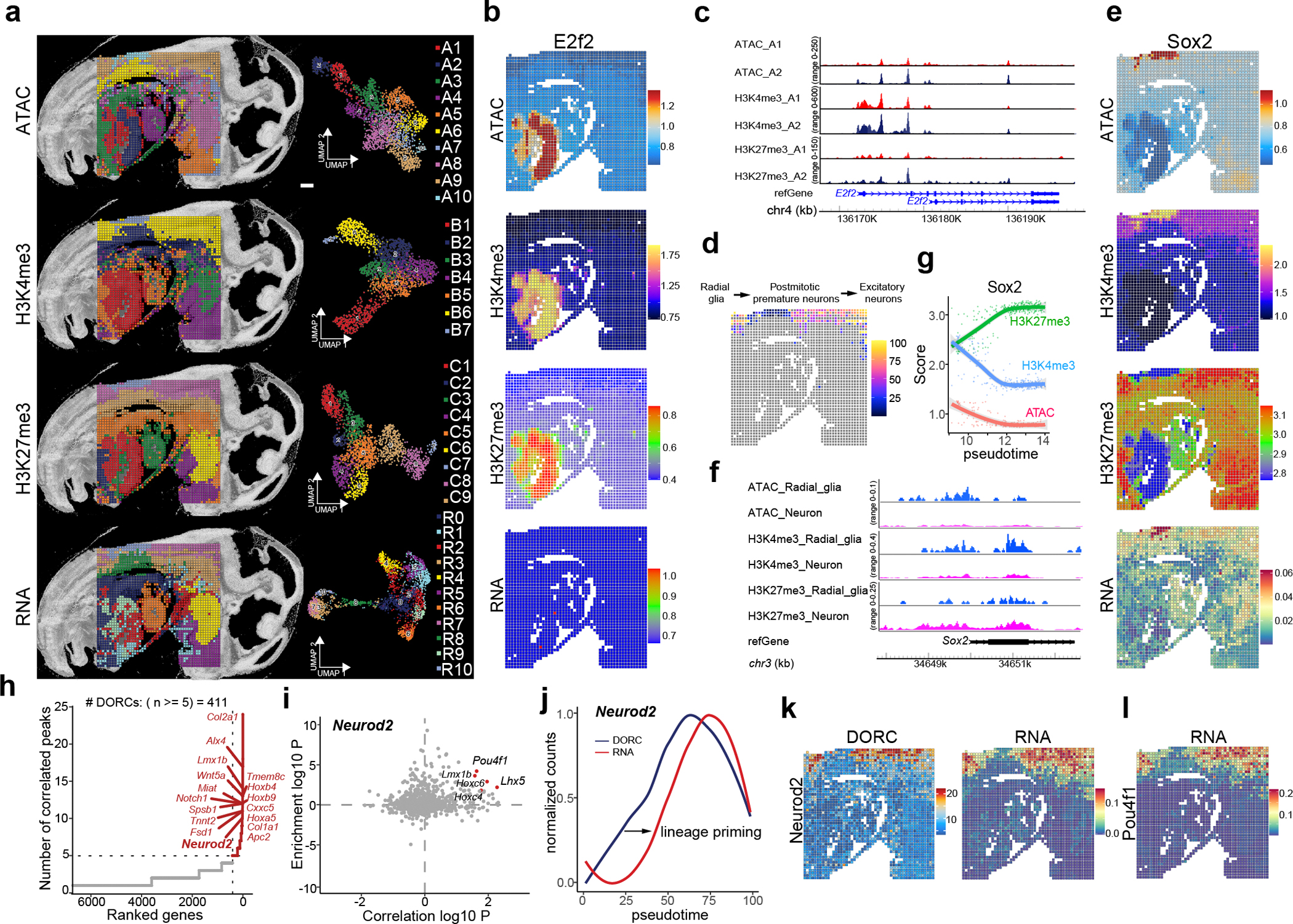
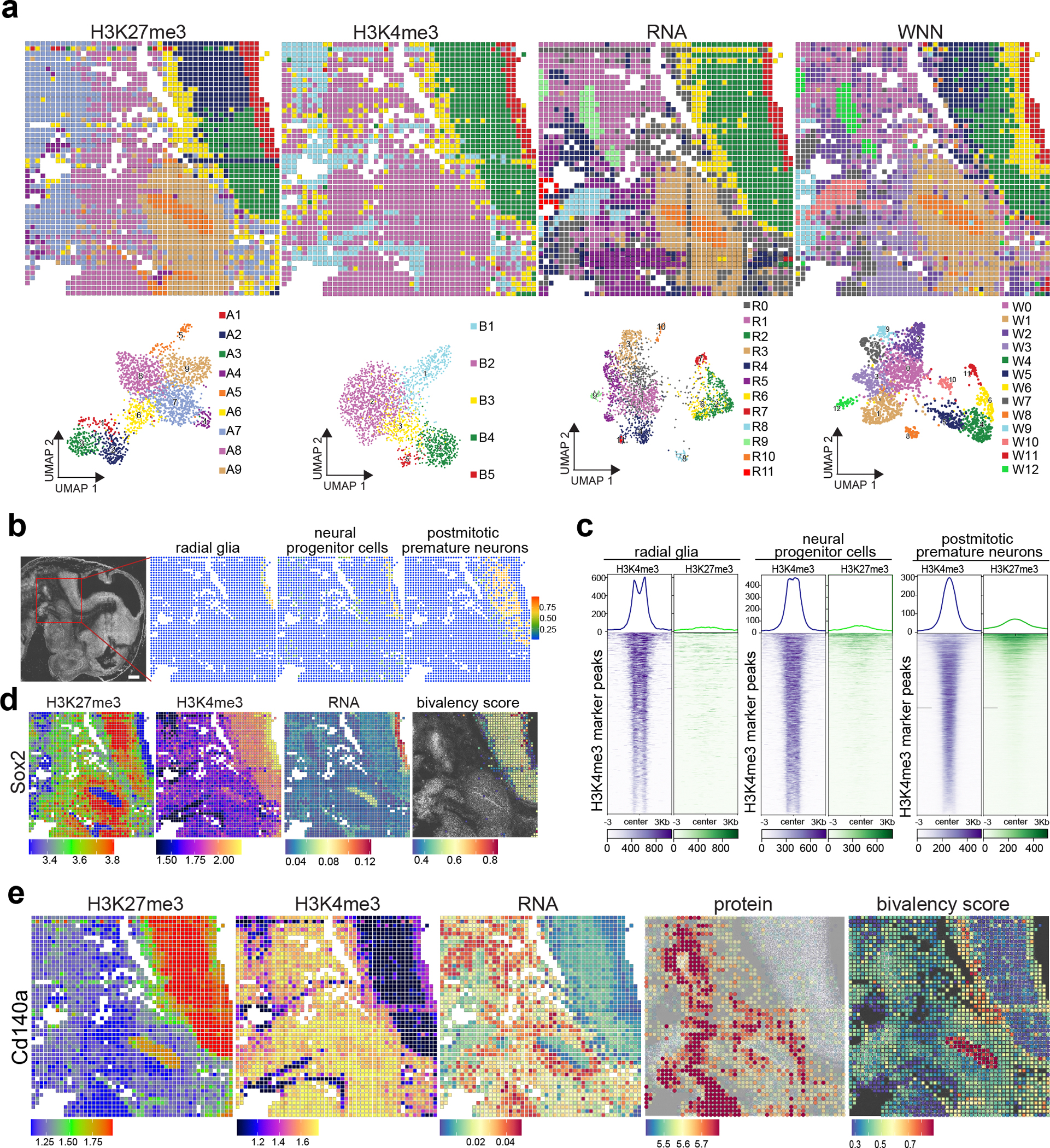
The phenotypic and functional states of cells are modulated by a complex interactive molecular hierarchy of multiple omics layers, involving the genome, epigenome, transcriptome, proteome and metabolome. Spatial omics approaches have enabled the study of these layers in tissue context but are often limited to one or two modalities, offering an incomplete view of cellular identity. Here we present spatial-Mux-seq, a multimodal spatial technology that allows simultaneous profiling of five different modalities: two histone modifications, chromatin accessibility, whole transcriptome and a panel of proteins at tissue scale and cellular level in a spatially resolved manner. We applied this technology to mouse embryos and mouse brains, generating detailed multimodal tissue maps that identified more cell types and states compared to unimodal data. This analysis uncovered spatiotemporal relationships among histone modifications, chromatin accessibility, gene expression and protein levels during neuron differentiation, and revealed a radial glia niche with spatially dynamic epigenetic signals. Collectively, the spatial multi-omics approach heralds a new era for characterizing tissue and cellular heterogeneity that single-modality studies alone could not reveal.
© 2025. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests: Y.D. and P.G. are inventors of a patent application related to this work. Y.D. is the scientific advisor of AtlasXomics Inc. M.L. receives research funding from Biogen Inc. unrelated to the current paper and is a cofounder of OmicPath AI LLC. The other authors declare no competing interests.
Figures










Update of
-
Multiplexed spatial mapping of chromatin features, transcriptome, and proteins in tissues.bioRxiv [Preprint]. 2024 Sep 19:2024.09.13.612892. doi: 10.1101/2024.09.13.612892. bioRxiv. 2024. Update in: Nat Methods. 2025 Mar;22(3):520-529. doi: 10.1038/s41592-024-02576-0. PMID: 39345645 Free PMC article. Updated. Preprint.
References
-
- Deng Y, Bai Z & Fan R Microtechnologies for single-cell and spatial multi-omics. Nature Reviews Bioengineering 1, 769–784 (2023). 10.1038/s44222-023-00084-y - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
