

Telomemore enables single-cell analysis of cell cycle and chromatin condensation
- PMID: 39878215
- PMCID: PMC11775621
- DOI: 10.1093/nar/gkaf031
Telomemore enables single-cell analysis of cell cycle and chromatin condensation
Abstract
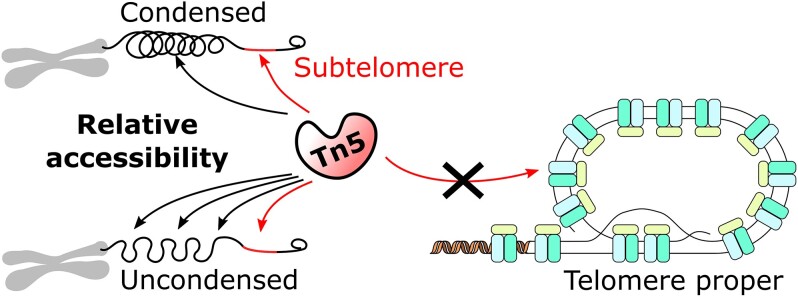
Single-cell RNA-seq methods can be used to delineate cell types and states at unprecedented resolution but do little to explain why certain genes are expressed. Single-cell ATAC-seq and multiome (ATAC + RNA) have emerged to give a complementary view of the cell state. It is however unclear what additional information can be extracted from ATAC-seq data besides transcription factor binding sites. Here, we show that ATAC-seq telomere-like reads counter-inituively cannot be used to infer telomere length, as they mostly originate from the subtelomere, but can be used as a biomarker for chromatin condensation. Using long-read sequencing, we further show that modern hyperactive Tn5 does not duplicate 9 bp of its target sequence, contrary to common belief. We provide a new tool, Telomemore, which can quantify nonaligning subtelomeric reads. By analyzing several public datasets and generating new multiome fibroblast and B-cell atlases, we show how this new readout can aid single-cell data interpretation. We show how drivers of condensation processes can be inferred, and how it complements common RNA-seq-based cell cycle inference, which fails for monocytes. Telomemore-based analysis of the condensation state is thus a valuable complement to the single-cell analysis toolbox.
© The Author(s) 2025. Published by Oxford University Press on behalf of Nucleic Acids Research.
Conflict of interest statement
J.T. is employed at the Sartorius. I.S.M. is employed at Umeå University but partially funded by the Sartorius. Other authors declare no conflict of interest.
Figures







Similar articles
-
Assessing the comparative effects of interventions in COPD: a tutorial on network meta-analysis for clinicians.Respir Res. 2024 Dec 21;25(1):438. doi: 10.1186/s12931-024-03056-x. Respir Res. 2024. PMID: 39709425 Free PMC article. Review.
-
Benchmarking algorithms for joint integration of unpaired and paired single-cell RNA-seq and ATAC-seq data.Genome Biol. 2023 Oct 24;24(1):244. doi: 10.1186/s13059-023-03073-x. Genome Biol. 2023. PMID: 37875977 Free PMC article.
-
scATAC-seq generates more accurate and complete regulatory maps than bulk ATAC-seq.Sci Rep. 2025 Jan 29;15(1):3665. doi: 10.1038/s41598-025-87351-7. Sci Rep. 2025. PMID: 39881144 Free PMC article.
-
Characterization of non-coding variants associated with transcription-factor binding through ATAC-seq-defined footprint QTLs in liver.Am J Hum Genet. 2025 Jun 5;112(6):1302-1315. doi: 10.1016/j.ajhg.2025.03.019. Epub 2025 Apr 17. Am J Hum Genet. 2025. PMID: 40250421
-
Defining disease severity in atopic dermatitis and psoriasis for the application to biomarker research: an interdisciplinary perspective.Br J Dermatol. 2024 Jun 20;191(1):14-23. doi: 10.1093/bjd/ljae080. Br J Dermatol. 2024. PMID: 38419411 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
