

Novel De Novo Intronic Variant of SYNGAP1 Associated With the Neurodevelopmental Disorders
- PMID: 39878419
- PMCID: PMC11775916
- DOI: 10.1002/mgg3.70066
Novel De Novo Intronic Variant of SYNGAP1 Associated With the Neurodevelopmental Disorders
Abstract
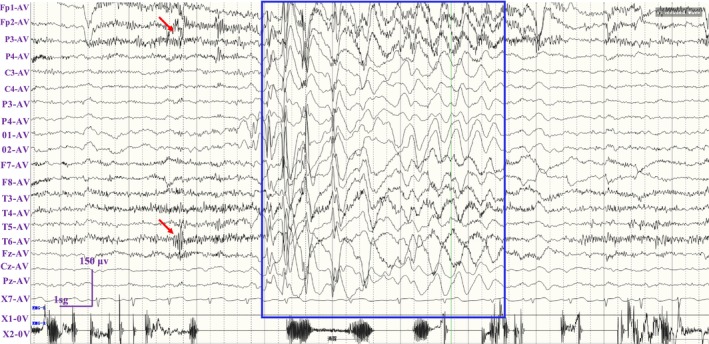
Background: SYNGAP1 encodes a Ras/Rap GTPase-activating protein that is predominantly expressed in the brain with the functional roles in regulating synaptic plasticity, spine morphogenesis, and cognition function. Pathogenic variants in SYNGAP1 have been associated with a spectrum of neurodevelopmental disorders characterized by developmental delays, intellectual disabilities, epilepsy, hypotonia, and the features of autism spectrum disorder. The aim of this study was to identify a novel SYNGAP1 gene variant linked to neurodevelopmental disorders and to evaluate the pathogenicity of the detected variant.
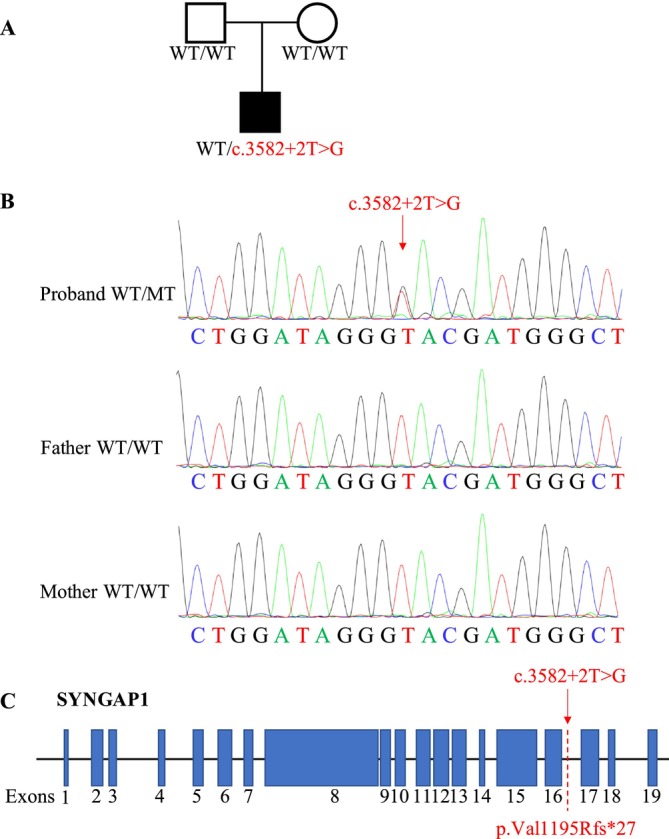
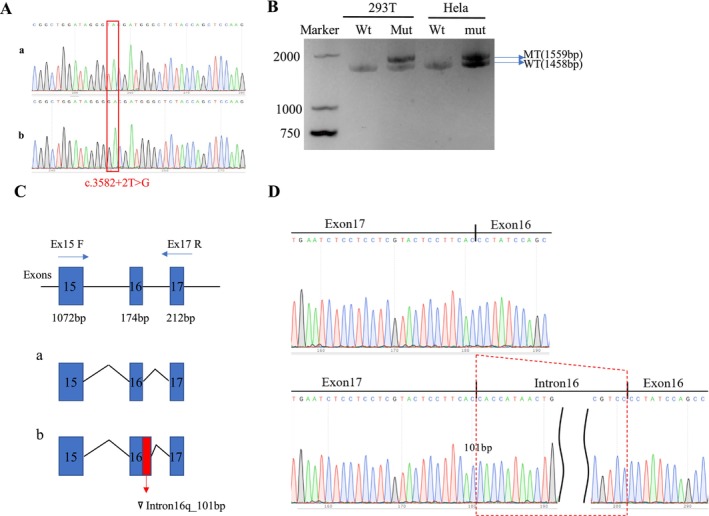
Methods: A novel de novo intronic variant in SYNGAP1 was identified by Whole exome sequencing (WES) and confirmed by Sanger sequencing. Minigene assays were conducted to assess whether the intronic variant in SYNGAP1 influenced the normal splicing of mRNA.
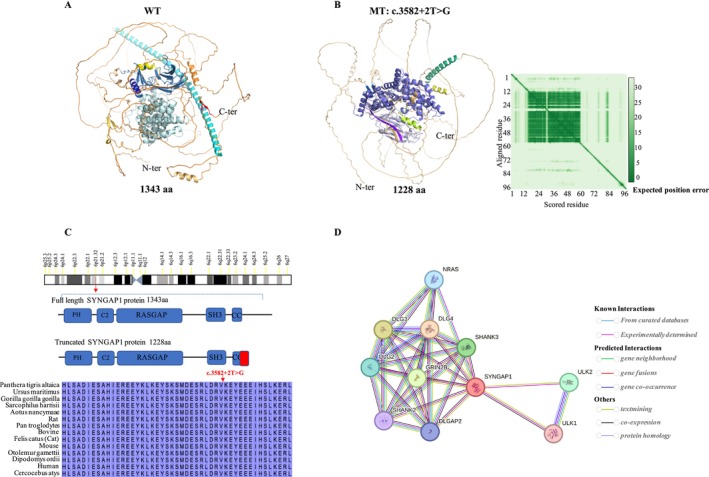
Results: A novel de novo intronic variant in SYNGAP1 (c.3582+2T>G) was indentified with clinical features suggestive of neurodevelopmental related disorders. Minigene splicing analysis demonstrated that this noncanonical splice site variant led to the activation of a cryptic acceptor splice site. Consequently, 101 base pairs of intron 16 were aberrantly retained in the mRNA, leading to a frameshift. This frameshift resulted in the introduction of a premature stop codon (TGA) in the coding sequence and the production of a truncated SYNGAP1 protein, potentially leding to loss of function and subsequent disruption of its biological roles.
Conclusion: Our findings highlight the significance of de novo pathogenic SYNGAP1 variants at the intron 16/exon 17 junction in the SYNGAP1-related neurodevelopmental disorders, providing novel insights into the genetic basis and diagnosis of these disabilities.
Keywords: SYNGAP1; intronic variation; minigene; variant interpretation; whole exome sequencing.
© 2025 The Author(s). Molecular Genetics & Genomic Medicine published by Wiley Periodicals LLC.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Identification and functional characterization of de novo variant in the SYNGAP1 gene causing intellectual disability.Front Genet. 2023 Oct 19;14:1270175. doi: 10.3389/fgene.2023.1270175. eCollection 2023. Front Genet. 2023. PMID: 37928246 Free PMC article.
-
Clinical Transcriptome Sequencing Confirms Activation of a Cryptic Splice Site in Suspected SYNGAP1-Related Disorder.Mol Syndromol. 2019 Jan;9(6):295-299. doi: 10.1159/000492706. Epub 2018 Aug 28. Mol Syndromol. 2019. PMID: 30800045 Free PMC article.
-
Mouse models of SYNGAP1 -related intellectual disability.bioRxiv [Preprint]. 2023 May 26:2023.05.25.542312. doi: 10.1101/2023.05.25.542312. bioRxiv. 2023. Update in: Proc Natl Acad Sci U S A. 2023 Sep 12;120(37):e2308891120. doi: 10.1073/pnas.2308891120. PMID: 37293116 Free PMC article. Updated. Preprint.
-
Species-conserved SYNGAP1 phenotypes associated with neurodevelopmental disorders.Mol Cell Neurosci. 2018 Sep;91:140-150. doi: 10.1016/j.mcn.2018.03.008. Epub 2018 Mar 24. Mol Cell Neurosci. 2018. PMID: 29580901 Free PMC article. Review.
-
SYNGAP1 mutations: Clinical, genetic, and pathophysiological features.Int J Dev Neurosci. 2019 Nov;78:65-76. doi: 10.1016/j.ijdevneu.2019.08.003. Epub 2019 Aug 24. Int J Dev Neurosci. 2019. PMID: 31454529 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
