

Targeting pancreatic cancer glutamine dependency confers vulnerability to GPX4-dependent ferroptosis
- PMID: 39879992
- PMCID: PMC11866519
- DOI: 10.1016/j.xcrm.2025.101928
Targeting pancreatic cancer glutamine dependency confers vulnerability to GPX4-dependent ferroptosis
Abstract
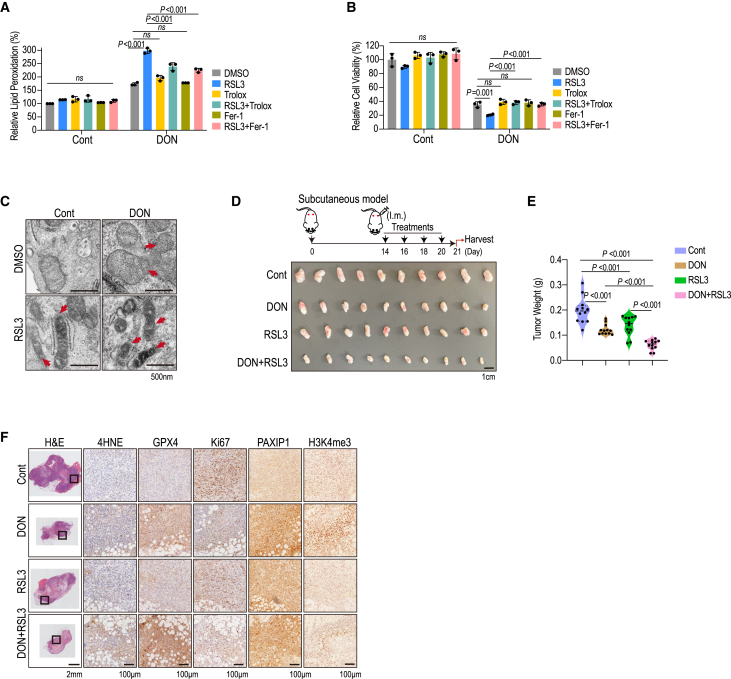
Pancreatic ductal adenocarcinoma (PDAC) relies heavily on glutamine (Gln) utilization to meet its metabolic and biosynthetic needs. How epigenetic regulators contribute to the metabolic flexibility and PDAC's response and adaptation to Gln scarcity in the tumor milieu remains largely unknown. Here, we elucidate that prolonged Gln restriction or treatment with the Gln antagonist, 6-diazo-5-oxo-L-norleucine (DON), leads to growth inhibition and ferroptosis program activation in PDAC. A CRISPR-Cas9 screen identifies an epigenetic regulator, Paxip1, which promotes H3K4me3 upregulation and Hmox1 transcription upon DON treatment. Additionally, ferroptosis-related repressors (e.g., Slc7a11 and Gpx4) are increased as an adaptive response, thereby predisposing PDAC cells to ferroptosis upon Gln deprivation. Moreover, DON sensitizes PDAC cells to GPX4 inhibitor-induced ferroptosis, both in vitro and in patient-derived xenografts (PDXs). Taken together, our findings reveal that targeting Gln dependency confers susceptibility to GPX4-dependent ferroptosis via epigenetic remodeling and provides a combination strategy for PDAC therapy.
Keywords: PDAC; combination therapy; epigenetic remodeling; ferroptosis; pancreatic ductal adenocarcinoma; prolonged glutamine starvation.
Copyright © 2025 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures







References
-
- Kamphorst J.J., Nofal M., Commisso C., Hackett S.R., Lu W., Grabocka E., Vander Heiden M.G., Miller G., Drebin J.A., Bar-Sagi D., et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75:544–553. doi: 10.1158/0008-5472.CAN-14-2211. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
