

Bat genomes illuminate adaptations to viral tolerance and disease resistance
- PMID: 39880942
- PMCID: PMC11821529
- DOI: 10.1038/s41586-024-08471-0
Bat genomes illuminate adaptations to viral tolerance and disease resistance
Abstract
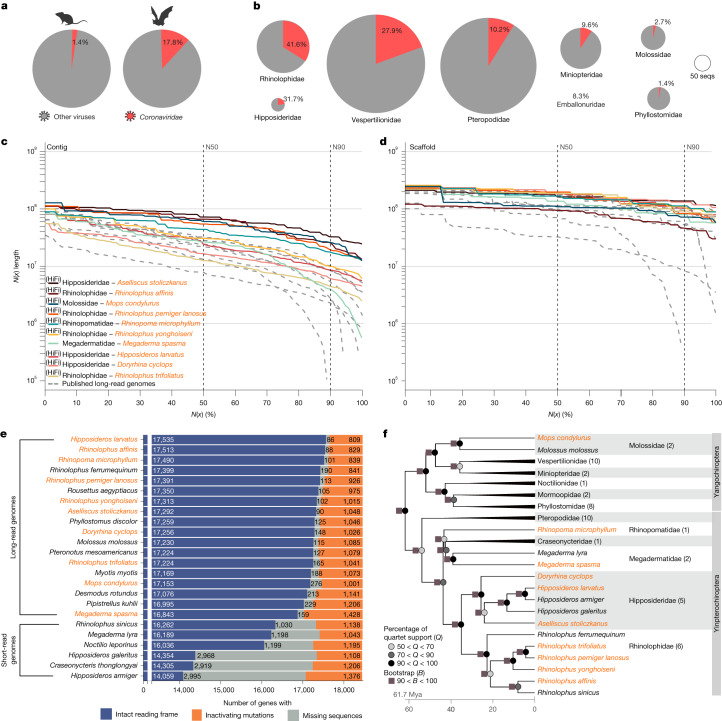
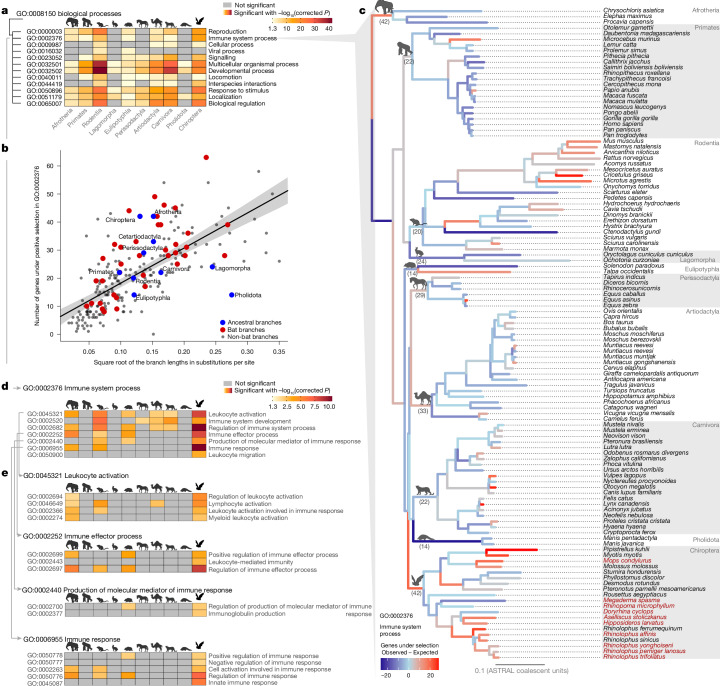
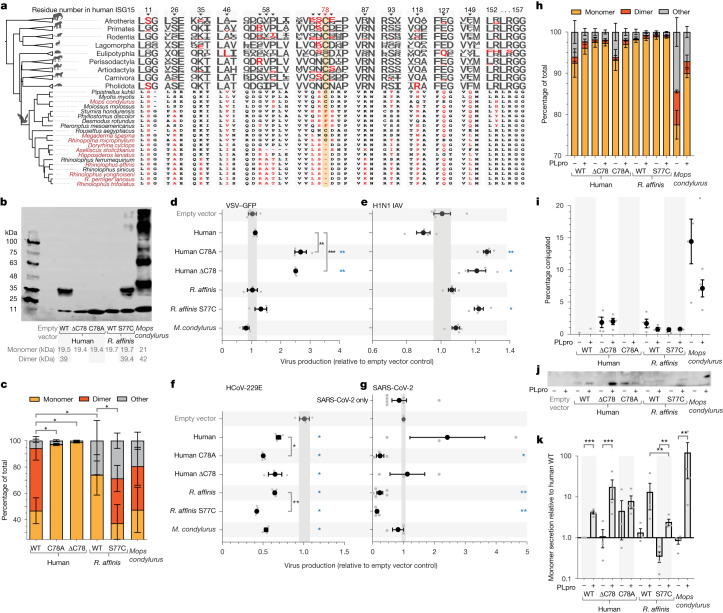
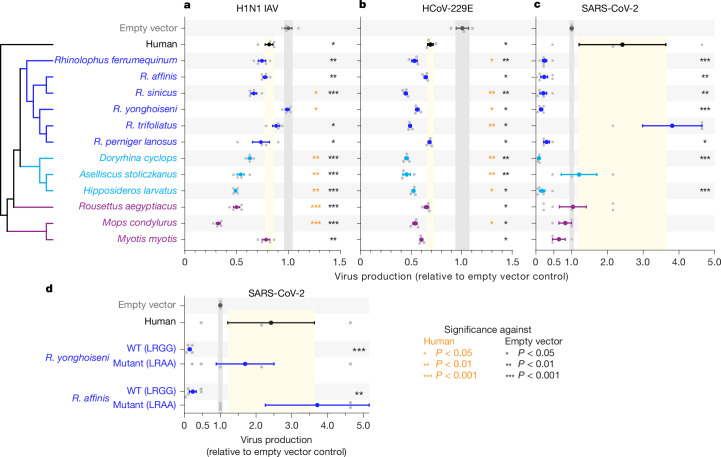
Zoonoses are infectious diseases transmitted from animals to humans. Bats have been suggested to harbour more zoonotic viruses than any other mammalian order1. Infections in bats are largely asymptomatic2,3, indicating limited tissue-damaging inflammation and immunopathology. To investigate the genomic basis of disease resistance, the Bat1K project generated reference-quality genomes of ten bat species, including potential viral reservoirs. Here we describe a systematic analysis covering 115 mammalian genomes that revealed that signatures of selection in immune genes are more prevalent in bats than in other mammalian orders. We found an excess of immune gene adaptations in the ancestral chiropteran branch and in many descending bat lineages, highlighting viral entry and detection factors, and regulators of antiviral and inflammatory responses. ISG15, which is an antiviral gene contributing to hyperinflammation during COVID-19 (refs. 4,5), exhibits key residue changes in rhinolophid and hipposiderid bats. Cellular infection experiments show species-specific antiviral differences and an essential role of protein conjugation in antiviral function of bat ISG15, separate from its role in secretion and inflammation in humans. Furthermore, in contrast to humans, ISG15 in most rhinolophid and hipposiderid bats has strong anti-SARS-CoV-2 activity. Our work reveals molecular mechanisms that contribute to viral tolerance and disease resistance in bats.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures











References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
