

SARS-CoV-2 evolution on a dynamic immune landscape
- PMID: 39880955
- PMCID: PMC11882442
- DOI: 10.1038/s41586-024-08477-8
SARS-CoV-2 evolution on a dynamic immune landscape
Abstract
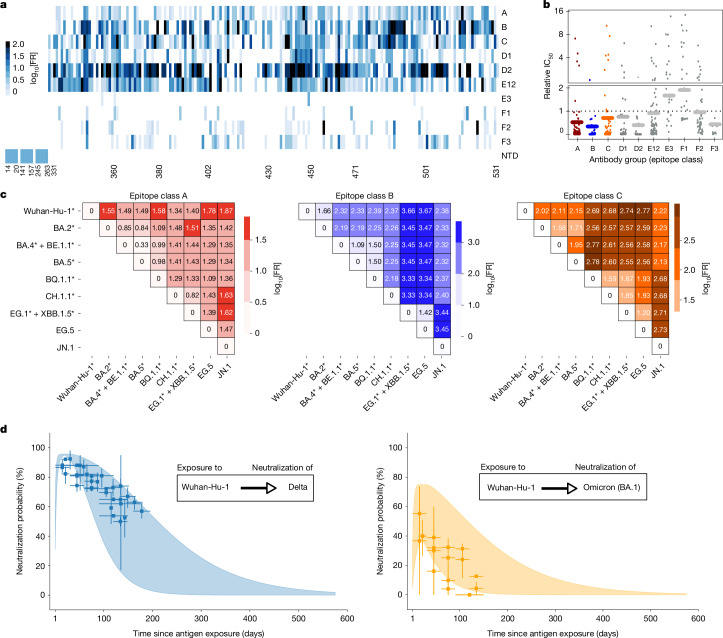
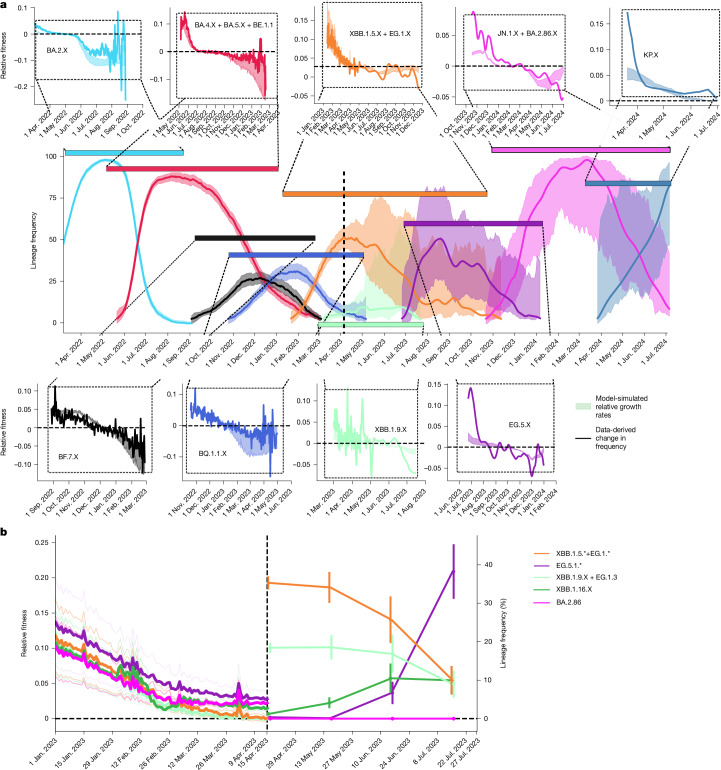
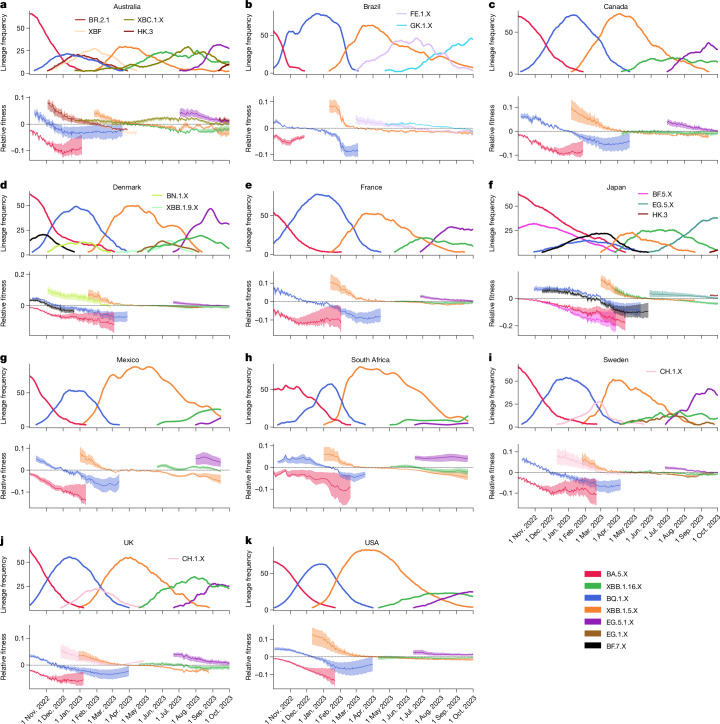
Since the onset of the pandemic, many SARS-CoV-2 variants have emerged, exhibiting substantial evolution in the virus' spike protein1, the main target of neutralizing antibodies2. A plausible hypothesis proposes that the virus evolves to evade antibody-mediated neutralization (vaccine- or infection-induced) to maximize its ability to infect an immunologically experienced population1,3. Because viral infection induces neutralizing antibodies, viral evolution may thus navigate on a dynamic immune landscape that is shaped by local infection history. Here we developed a comprehensive mechanistic model, incorporating deep mutational scanning data4,5, antibody pharmacokinetics and regional genomic surveillance data, to predict the variant-specific relative number of susceptible individuals over time. We show that this quantity precisely matched historical variant dynamics, predicted future variant dynamics and explained global differences in variant dynamics. Our work strongly suggests that the ongoing pandemic continues to shape variant-specific population immunity, which determines a variant's ability to transmit, thus defining variant fitness. The model can be applied to any region by utilizing local genomic surveillance data, allows contextualizing risk assessment of variants and provides information for vaccine design.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures












References
-
- Markov, P. V. et al. The evolution of SARS-CoV-2. Nat. Rev. Microbiol.21, 361–379 (2023). - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
