

Integrating single-cell RNA and T cell/B cell receptor sequencing with mass cytometry reveals dynamic trajectories of human peripheral immune cells from birth to old age
- PMID: 39881000
- PMCID: PMC11785523
- DOI: 10.1038/s41590-024-02059-6
Integrating single-cell RNA and T cell/B cell receptor sequencing with mass cytometry reveals dynamic trajectories of human peripheral immune cells from birth to old age
Abstract
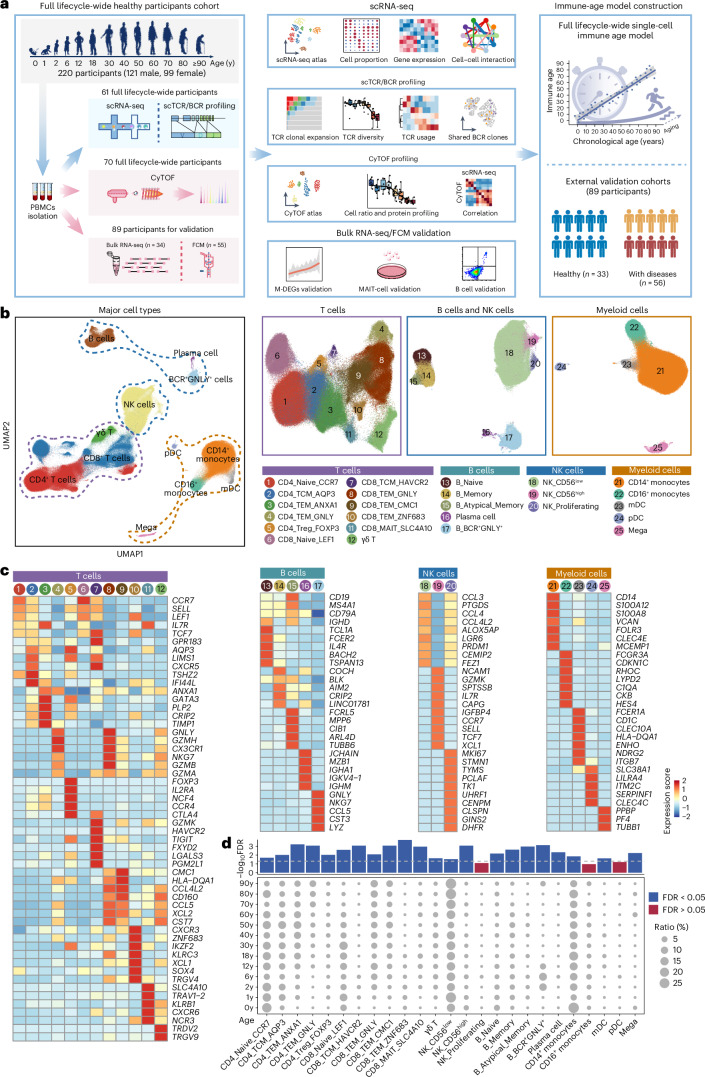
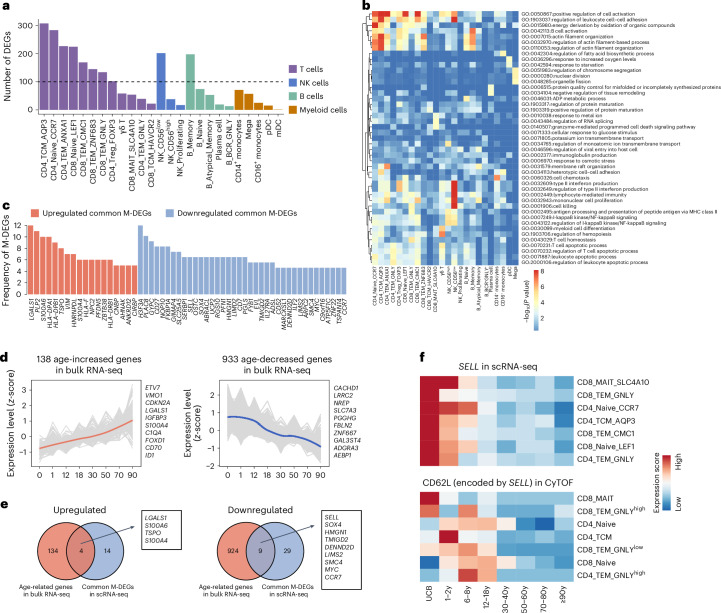
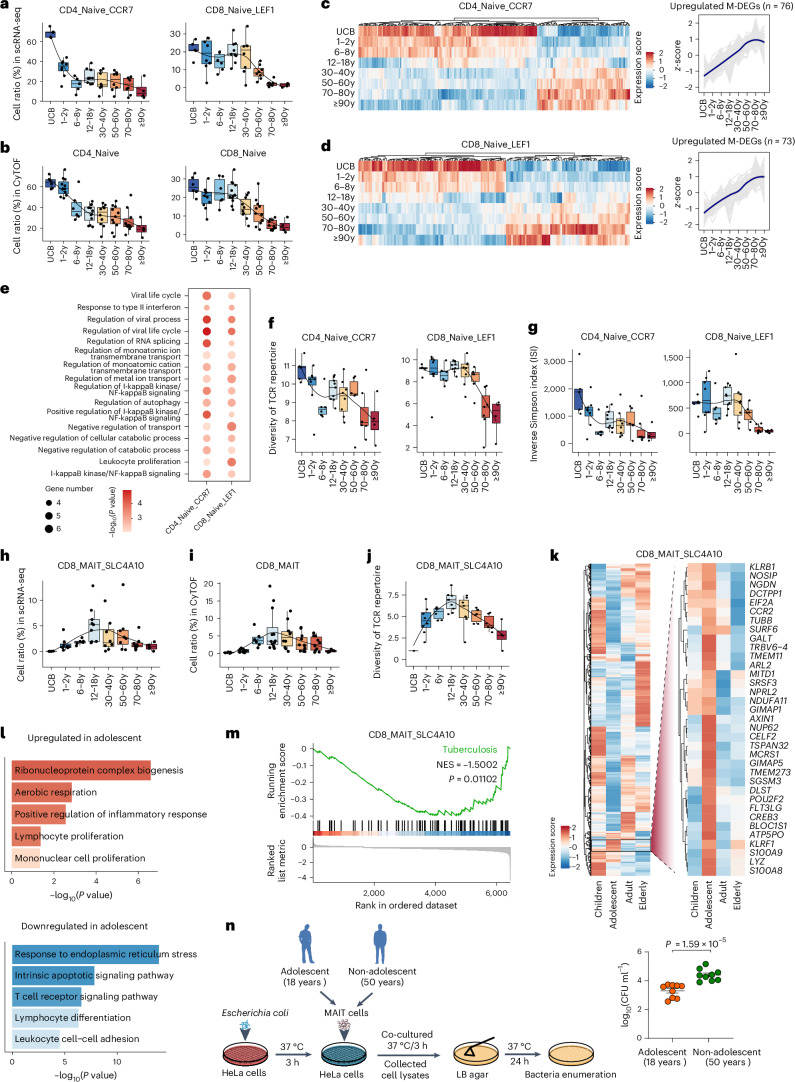
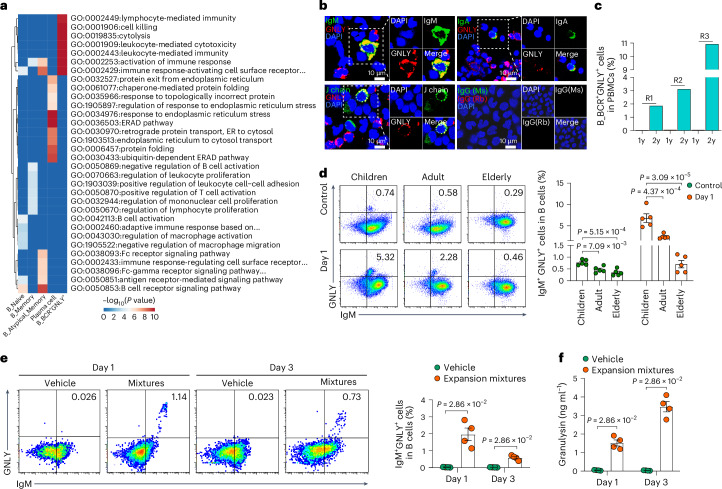
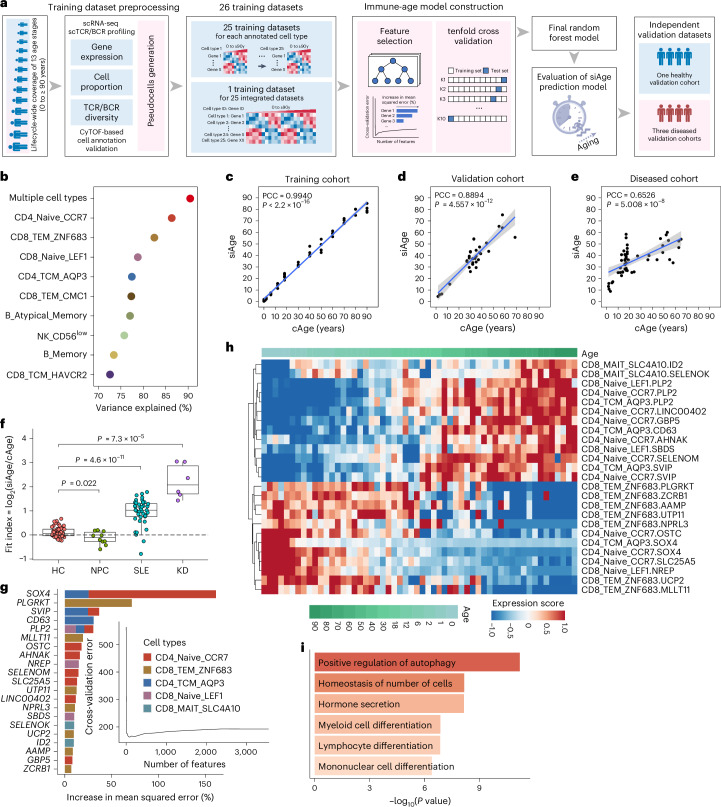
A comprehensive understanding of the evolution of the immune landscape in humans across the entire lifespan at single-cell transcriptional and protein levels, during development, maturation and senescence is currently lacking. We recruited a total of 220 healthy volunteers from the Shanghai Pudong Cohort (NCT05206643), spanning 13 age groups from 0 to over 90 years, and profiled their peripheral immune cells through single-cell RNA-sequencing coupled with single T cell and B cell receptor sequencing, high-throughput mass cytometry, bulk RNA-sequencing and flow cytometry validation experiments. We revealed that T cells were the most strongly affected by age and experienced the most intensive rewiring in cell-cell interactions during specific age. Different T cell subsets displayed different aging patterns in both transcriptomes and immune repertoires; examples included GNLY+CD8+ effector memory T cells, which exhibited the highest clonal expansion among all T cell subsets and displayed distinct functional signatures in children and the elderly; and CD8+ MAIT cells, which reached their peaks of relative abundance, clonal diversity and antibacterial capability in adolescents and then gradually tapered off. Interestingly, we identified and experimentally verified a previously unrecognized 'cytotoxic' B cell subset that was enriched in children. Finally, an immune age prediction model was developed based on lifecycle-wide single-cell data that can evaluate the immune status of healthy individuals and identify those with disturbed immune functions. Our work provides both valuable insights and resources for further understanding the aging of the immune system across the whole human lifespan.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures












References
-
- Mittelbrunn, M. & Kroemer, G. Hallmarks of T cell aging. Nat. Immunol.22, 687–698 (2021). - PubMed
-
- Tong, Z. et al. Single-cell RNA sequencing maps immune cell heterogeneity in mice with allogeneic cardiac transplantation. Cardiovasc. Innov. Appl.8, 988 (2023).
-
- Nikolich-Zugich, J. The twilight of immunity: emerging concepts in aging of the immune system. Nat. Immunol.19, 10–19 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
