

Growth hormone-releasing hormone signaling and manifestations within the cardiovascular system
- PMID: 39883351
- PMCID: PMC12137388
- DOI: 10.1007/s11154-024-09939-0
Growth hormone-releasing hormone signaling and manifestations within the cardiovascular system
Abstract
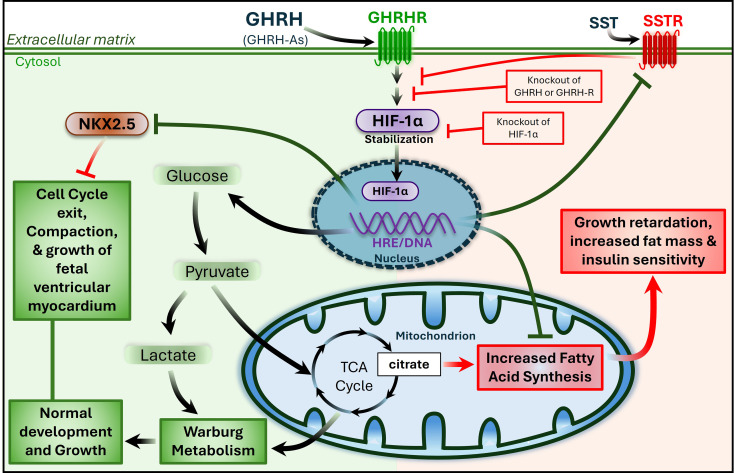
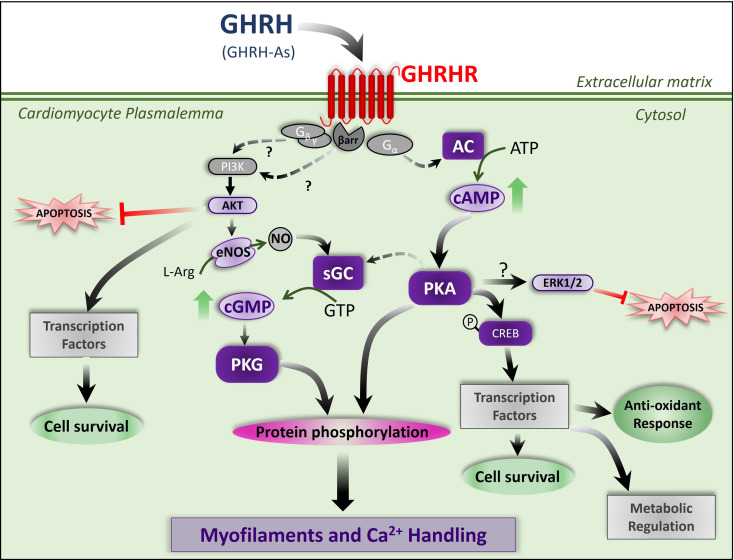
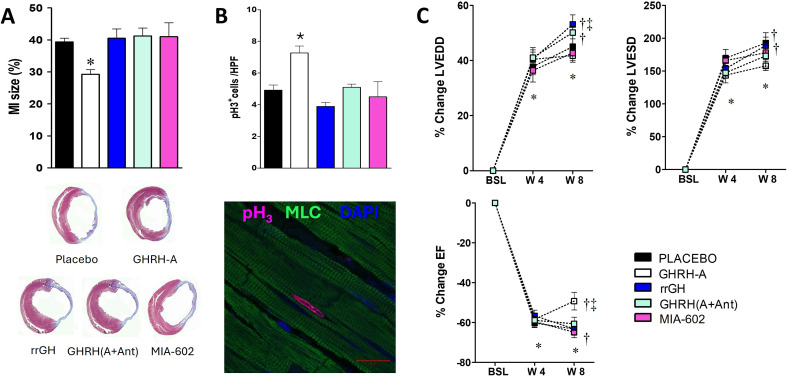
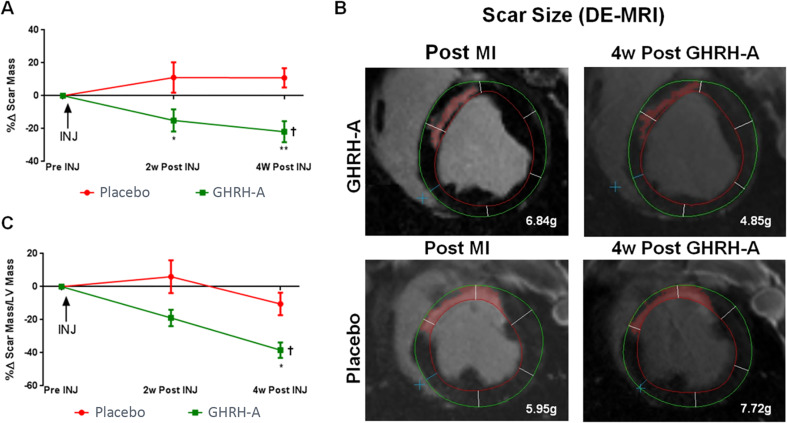
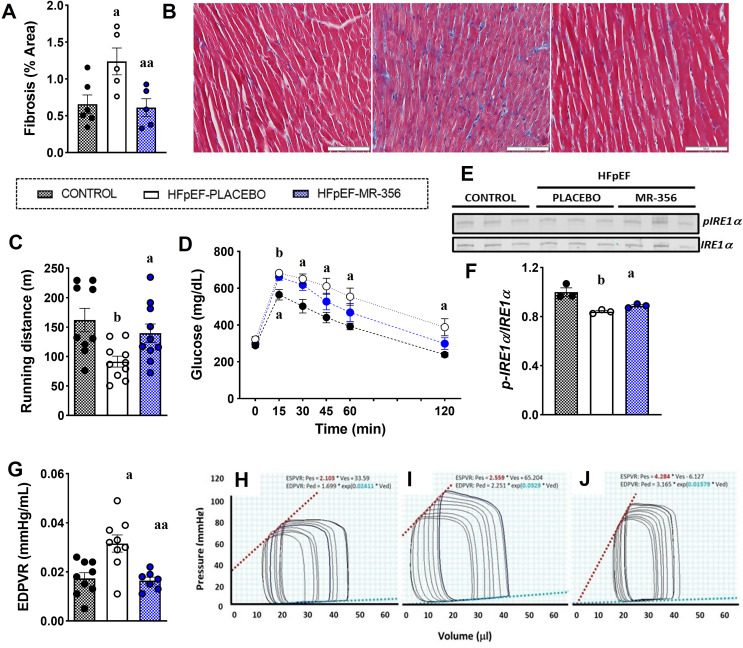
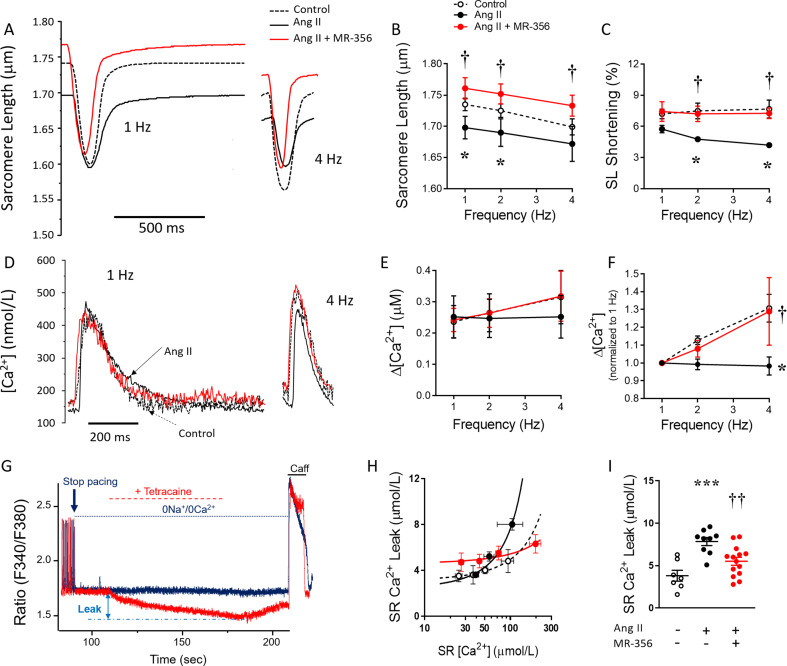
Growth hormone (GH)-releasing hormone (GHRH), a hypothalamic peptide initially characterized for its role in GH regulation, has gained increasing attention due to its GH-independent action on peripheral physiology, including that of the cardiovascular system. While its effects on the peripheral vasculature are still under investigation, GHRH and synthetic agonists have exhibited remarkable receptor-mediated cardioprotective properties in preclinical models. GHRH and its analogs enhance myocardial function by improving contractility, reducing oxidative stress, inflammation, and offsetting pathological remodeling. Studies performed in small and large animal models have demonstrated the efficacy of these compounds in diverse cardiomyopathies, suggesting their potential as promising therapeutic agents. However, the clinical translation of GHRH synthetic analogs still faces challenges related to the route of administration and potential side effects mainly associated with activation of the GH/IGF-I axis. Despite these hurdles, the compelling evidence supporting their role in cardiac repair makes GHRH analogs attractive candidates for clinical testing in the treatment of various cardiac diseases.
Keywords: Cardioprotective; Cardiovascular; GHRH receptors; GHRH-analogs; Growth hormone-releasing hormone.
© 2024. The Author(s).
Conflict of interest statement
Declarations. Competing interests: R.M.KT. and J.M.H. are listed as co-inventors on patents on GHRH analogues which were assigned to the University of Miami and Veterans Affairs Department. J.M.H. previously owned equity in Biscayne Pharmaceuticals, license of intellectual property used in this study. Biscayne Pharmaceuticals did not provide funding for this study. JMH reported having a patent for cardiac cell-based therapy. He holds equity in Vestion Inc. and maintains a professional relationship with Vestion Inc. as a consultant and member of the Board of Directors and Scientific Advisory Board. Dr. Joshua Hare is the Chief Scientific Officer, a compensated consultant and advisory board member for Longeveron, and holds equity in Longeveron. Dr. Hare is also the co-inventor of intellectual property licensed to Longeveron. The University of Miami also stands to gain royalties from the commercialization of the IP.K.E.H. holds equity in Vestion Inc. He is also the co-inventor of intellectual property licensed to Vestion Inc and Longeveron. K.E.H. is the Chief Executive Officer for KosBio P.C. and holds equity in KosBio. He is also the co-inventor of intellectual property related to GHRH/GHRHR signaling, described in this work. Longeveron LLC, Vestion Inc. and KosBio P.C. did not participate in funding this work. All other authors have declared that no conflict of interest exists.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
