

Lactylation of HDAC1 Confers Resistance to Ferroptosis in Colorectal Cancer
- PMID: 39888307
- PMCID: PMC11947995
- DOI: 10.1002/advs.202408845
Lactylation of HDAC1 Confers Resistance to Ferroptosis in Colorectal Cancer
Abstract
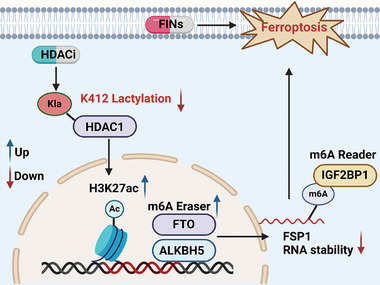
Colorectal cancer (CRC) is highly resistant to ferroptosis, which hinders the application of anti-ferroptosis therapy. Through drug screening, it is found that histone deacetylase inhibitor (HDACi) significantly sensitized CRC to ferroptosis. The combination of HDACi and ferroptosis inducers synergically suppresses CRC growth both in vivo and in vitro. Mechanically, HDACi reduces ferroptosis suppressor protein (FSP1) by promoting its mRNA degradation. Specifically, it is confirmed that HDACi specifically targets HDAC1 and promotes the H3K27ac modification of fat mass- and obesity-associated gene (FTO) and AlkB Homolog 5, RNA Demethylase (ALKBH5), which results in significant activation of FTO and ALKBH5. The activation of FTO and ALKBH5 reduces N6-methyladenosine (m6A) modification on FSP1 mRNA, leading to its degradation. Crucially, lactylation of HDAC1K412 is essential for ferroptosis regulation. Both Vorinostat (SAHA) and Trichostatin A (TSA) notably diminish HDAC1K412 lactylation in comparison to other HDAC1 inhibitors, exhibiting a consistent trend of increasing susceptibility to ferroptosis. In conclusion, the research reveals that HDACi decreases HDAC1K412 lactylation to sensitize CRC to ferroptosis and that the combination of HDACi and ferroptosis inducers can be a promising therapeutic strategy for CRC.
Keywords: HDAC inhibitor; N6‐methyladenosine modification; colorectal cancer; ferroptosis; lactylation.
© 2025 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
-
- a) Zou Y., Palte M. J., Deik A. A., Li H., Eaton J. K., Wang W., Tseng Y. Y., Deasy R., Kost‐Alimova M., Dančík V., Leshchiner E. S., Viswanathan V. S., Signoretti S., Choueiri T. K., Boehm J. S., Wagner B. K., Doench J. G., Clish C. B., Clemons P. A., Schreiber S. L., Nat. Commun. 2019, 10, 1617; - PMC - PubMed
- b) Koppula P., Lei G., Zhang Y., Yan Y., Mao C., Kondiparthi L., Shi J., Liu X., Horbath A., Das M., Nat. Commun. 2022, 13, 2206; - PMC - PubMed
- c) Magri J., Gasparetto A., Conti L., Calautti E., Cossu C., Ruiu R., Barutello G., Cavallo F., Cells 2021, 10, 108. - PMC - PubMed
-
- Bondarev A. D., Attwood M. M., Jonsson J., Chubarev V. N., Tarasov V. V., Schiöth H. B., Br. J. Clin. Pharmacol. 2021, 87, 4577. - PubMed
MeSH terms
Substances
Grants and funding
- 82372749/Natural Science Foundation of China
- 82303206/Natural Science Foundation of China
- 82072951/Natural Science Foundation of China
- 20Y11914300/Science and Technology Commission of Shanghai Municipality
- 22Y21900100/Science and Technology Commission of Shanghai Municipality
- SACA-AX202213/Shanghai Anticancer Association
- BK20231261/Natural Science Foundation of Jiangsu Province
- BK20231146/Natural Science Foundation of Jiangsu Province
- WMCG202326/The General Program of Wuxi Medical Center, Nanjing Medical University
- WMCG202326/The General Program of Wuxi Medical Center
- TZKY20230308/Major Research Projects of Taizhou Clinical Medical College
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous