

An integrative multiparametric approach stratifies putative distinct phenotypes of blast phase chronic myelomonocytic leukemia
- PMID: 39892394
- PMCID: PMC11866517
- DOI: 10.1016/j.xcrm.2025.101933
An integrative multiparametric approach stratifies putative distinct phenotypes of blast phase chronic myelomonocytic leukemia
Abstract
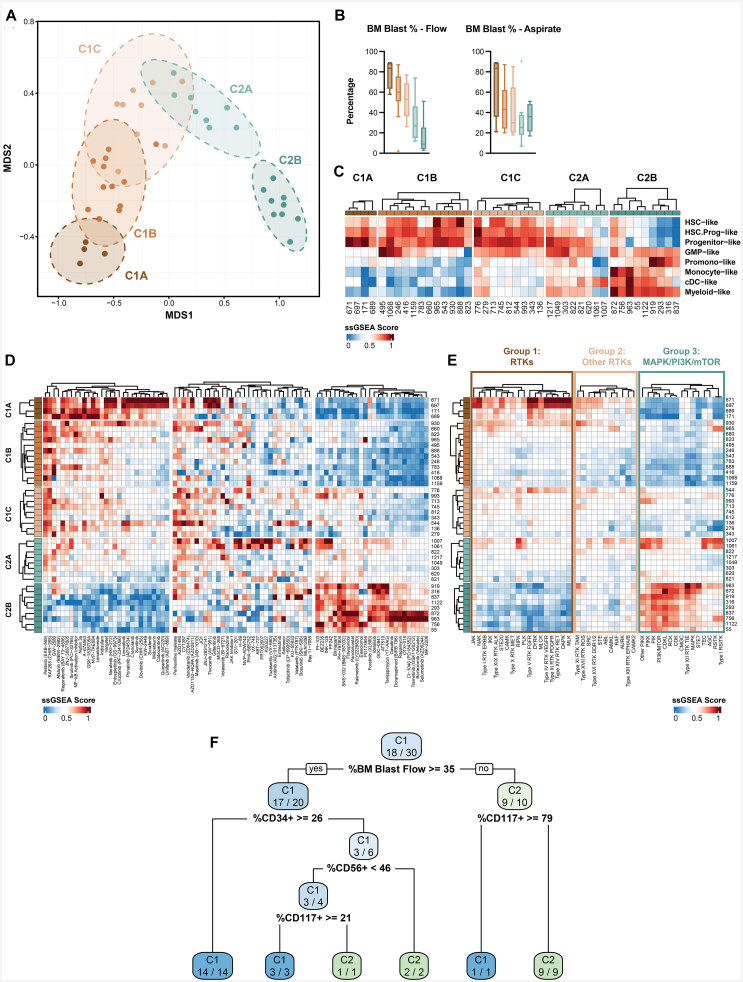
Approximately 30% of patients with chronic myelomonocytic leukemia (CMML) undergo transformation to a chemo-refractory blastic phase (BP-CMML). Seeking novel therapeutic approaches, we profiled blast transcriptomes from 42 BP-CMMLs, observing extensive transcriptional heterogeneity and poor alignment to current acute myeloid leukemia (AML) classifications. BP-CMMLs display distinctive transcriptomic profiles, including enrichment for quiescence and variability in drug response signatures. Integrating clinical, immunophenotype, and transcriptome parameters, Random Forest unsupervised clustering distinguishes immature and mature subtypes characterized by differential expression of transcriptional modules, oncogenes, apoptotic regulators, and patterns of surface marker expression. Subtypes differ in predicted response to AML drugs, validated ex vivo in primary samples. Iteratively refined stratification resolves a classification structure comprising five subtypes along a maturation spectrum, predictive of response to novel agents including consistent patterns for receptor tyrosine kinase (RTK), cyclin-dependent kinase (CDK), mechanistic target of rapamycin (MTOR), and mitogen-activated protein kinase (MAPK) inhibitors. Finally, we generate a prototype decision tree to stratify BP-CMML with high specificity and sensitivity, requiring validation but with potential clinical applicability to guide personalized drug selection for improved outcomes.
Keywords: CMML; MDS/MPN; blast phase; leukemic transformation; secondary AML.
Copyright © 2025. Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures







References
-
- Khoury J.D., Solary E., Abla O., Akkari Y., Alaggio R., Apperley J.F., Bejar R., Berti E., Busque L., Chan J.K.C., et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36:1703–1719. - PMC - PubMed
-
- Solary E., Itzykson R. How I treat chronic myelomonocytic leukemia. Blood. 2017;130:126–136. - PubMed
-
- Valent P., Orazi A., Savona M.R., Patnaik M.M., Onida F., van de Loosdrecht A.A., Haase D., Haferlach T., Elena C., Pleyer L., et al. Proposed diagnostic criteria for classical chronic myelomonocytic leukemia (CMML), CMML variants and pre-CMML conditions. Haematologica. 2019;104:1935–1949. - PMC - PubMed
-
- Batta K., Bossenbroek H.M., Pemmaraju N., Wilks D.P., Chasty R., Dennis M., Milne P., Collin M., Beird H.C., Taylor J., et al. Divergent clonal evolution of blastic plasmacytoid dendritic cell neoplasm and chronic myelomonocytic leukemia from a shared TET2-mutated origin. Leukemia. 2021;35:3299–3303. - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
