

Homeostasis and metabolism of iron and other metal ions in neurodegenerative diseases
- PMID: 39894843
- PMCID: PMC11788444
- DOI: 10.1038/s41392-024-02071-0
Homeostasis and metabolism of iron and other metal ions in neurodegenerative diseases
Abstract
As essential micronutrients, metal ions such as iron, manganese, copper, and zinc, are required for a wide range of physiological processes in the brain. However, an imbalance in metal ions, whether excessive or insufficient, is detrimental and can contribute to neuronal death through oxidative stress, ferroptosis, cuproptosis, cell senescence, or neuroinflammation. These processes have been found to be involved in the pathological mechanisms of neurodegenerative diseases. In this review, the research history and milestone events of studying metal ions, including iron, manganese, copper, and zinc in neurodegenerative diseases such as Parkinson's disease (PD), Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), and Huntington's disease (HD), will be introduced. Then, the upstream regulators, downstream effector, and crosstalk of mental ions under both physiologic and pathologic conditions will be summarized. Finally, the therapeutic effects of metal ion chelators, such as clioquinol, quercetin, curcumin, coumarin, and their derivatives for the treatment of neurodegenerative diseases will be discussed. Additionally, the promising results and limitations observed in clinical trials of these metal ion chelators will also be addressed. This review will not only provide a comprehensive understanding of the role of metal ions in disease development but also offer perspectives on their modulation for the prevention or treatment of neurodegenerative diseases.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures


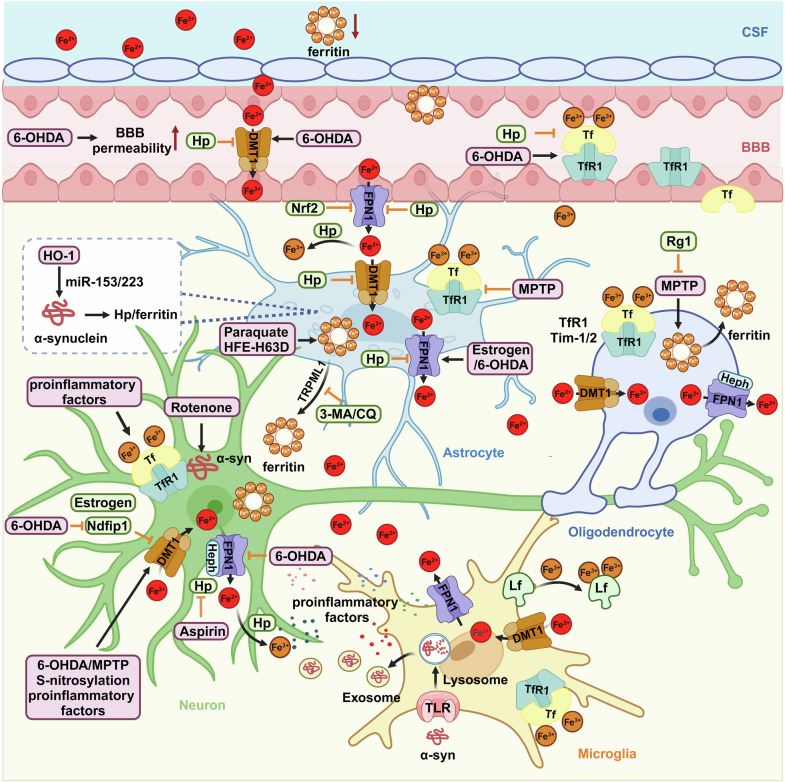
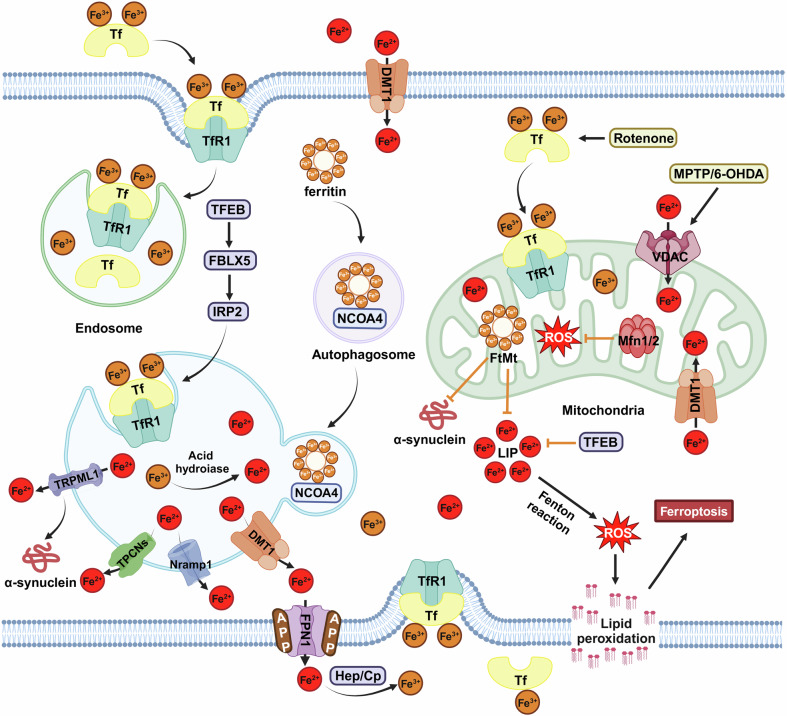

References
-
- Dexter, D. T. et al. Increased nigral iron content in postmortem parkinsonian brain. Lancet2, 1219–1220 (1987). - PubMed
-
- Dexter, D. T. et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain114, 1953–1975 (1991). - PubMed
-
- Dexter, D. T. et al. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem52, 1830–1836 (1989). - PubMed
-
- Mann, V. M. et al. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann. Neurol.36, 876–881 (1994). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 32471049/National Natural Science Foundation of China (National Science Foundation of China)
- 32170984/National Natural Science Foundation of China (National Science Foundation of China)
- 32200802/National Natural Science Foundation of China (National Science Foundation of China)
- ZR2020YQ23/Natural Science Foundation of Shandong Province (Shandong Provincial Natural Science Foundation)
- ZR2024MC153/Natural Science Foundation of Shandong Province (Shandong Provincial Natural Science Foundation)
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
