

GRHL2-HER3 and E-cadherin mediate EGFR-bypass drug resistance in lung cancer cells
- PMID: 39897079
- PMCID: PMC11782226
- DOI: 10.3389/fcell.2024.1511190
GRHL2-HER3 and E-cadherin mediate EGFR-bypass drug resistance in lung cancer cells
Abstract
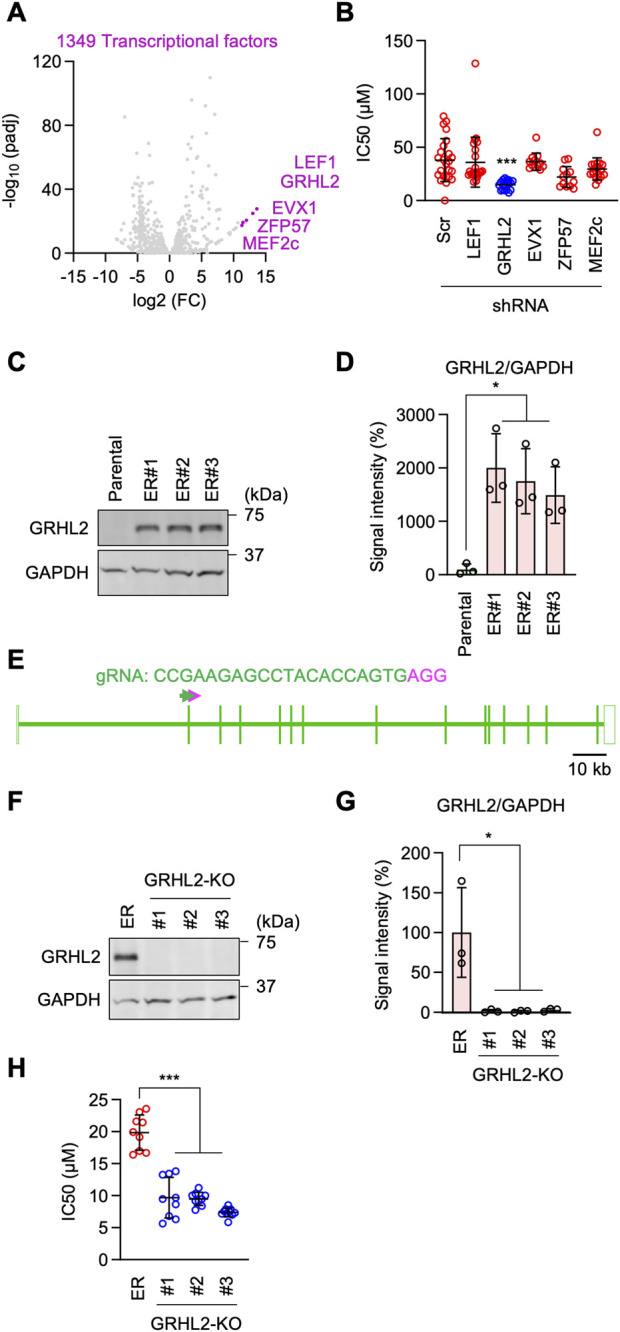
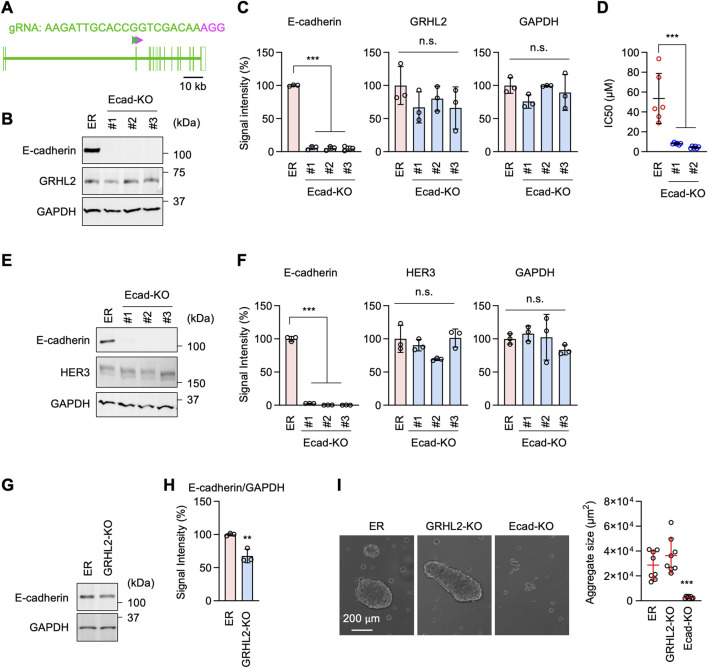
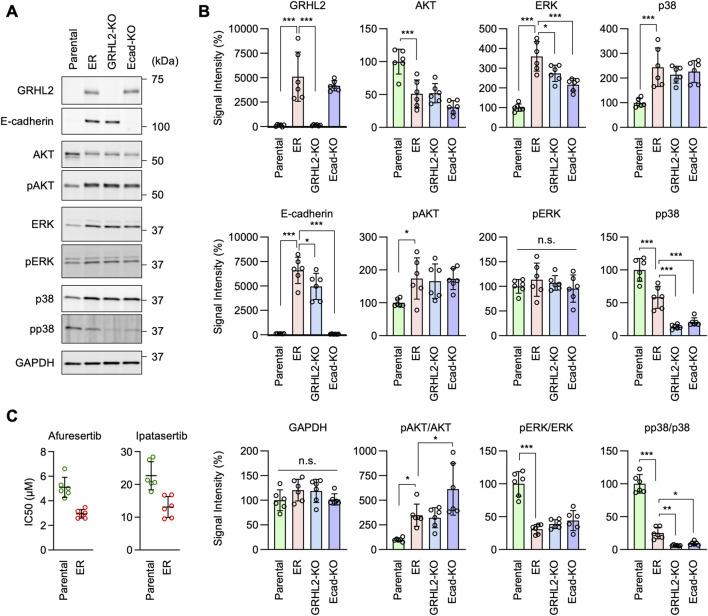
Epidermal growth factor receptor (EGFR) is a major oncogenic protein, and thus EGFR-targeting therapies are widely used in patients with various types of cancer, including lung cancer. However, resistance to EGFR inhibitors, such as erlotinib, presents a significant challenge in treating lung cancer. In this study, we established an EGFR-independent, erlotinib-resistant (ER) phenotype in lung cancer A549 cells by exposing them to erlotinib for an extended period. The resulting ER cells exhibited a dramatic increase in erlotinib resistance, a decreased EGFR protein level, and enhanced tumor growth, suggesting a robust mechanism bypassing EGFR inhibition. RNA sequencing identified the transcription factor GRHL2 as a critical player in this resistance. GRHL2 was upregulated in ER cells, and its knockdown and knockout significantly reduced erlotinib resistance. Further analysis revealed that GRHL2 upregulates the receptor tyrosine kinase HER3, and that HER3 knockdown similarly decreases the IC50 for erlotinib. Additionally, ER cells showed increased cell-cell adhesion, linked to upregulated E-cadherin. E-cadherin was found to be vital for erlotinib resistance, largely independent of GRHL2, highlighting multiple parallel pathways sustaining resistance. These findings provide a novel mechanism of drug resistance and suggest that combination therapies targeting both GRHL2-HER3 and E-cadherin-mediated pathways may be necessary to overcome erlotinib resistance in lung cancer.
Keywords: EGFR; GRHL2; HER3; cadherin; drug resistance; lung cancer.
Copyright © 2025 Ito, Iwata, Adachi, Sesaki and Iijima.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision
Figures






References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous