

Prediction of influenza A virus-human protein-protein interactions using XGBoost with continuous and discontinuous amino acids information
- PMID: 39897484
- PMCID: PMC11787804
- DOI: 10.7717/peerj.18863
Prediction of influenza A virus-human protein-protein interactions using XGBoost with continuous and discontinuous amino acids information
Abstract
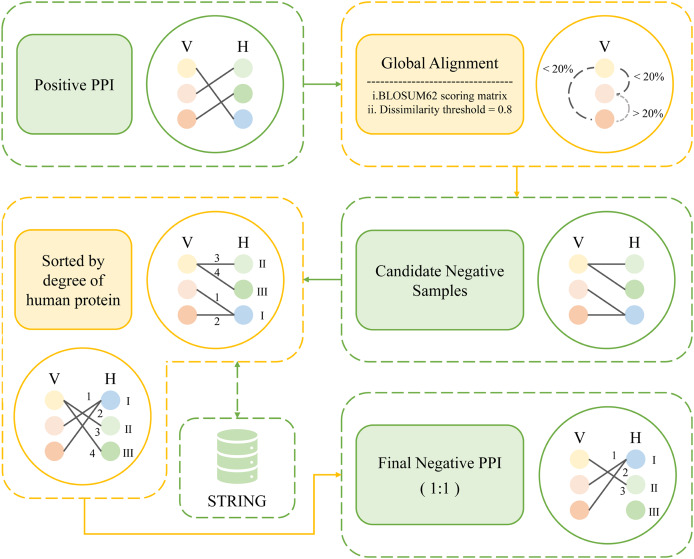
Influenza A virus (IAV) has the characteristics of high infectivity and high pathogenicity, which makes IAV infection a serious public health threat. Identifying protein-protein interactions (PPIs) between IAV and human proteins is beneficial for understanding the mechanism of viral infection and designing antiviral drugs. In this article, we developed a sequence-based machine learning method for predicting PPI. First, we applied a new negative sample construction method to establish a high-quality IAV-human PPI dataset. Then we used conjoint triad (CT) and Moran autocorrelation (Moran) to encode biologically relevant features. The joint consideration utilizing the complementary information between contiguous and discontinuous amino acids provides a more comprehensive description of PPI information. After comparing different machine learning models, the eXtreme Gradient Boosting (XGBoost) model was determined as the final model for the prediction. The model achieved an accuracy of 96.89%, precision of 98.79%, recall of 94.85%, F1-score of 96.78%. Finally, we successfully identified 3,269 potential target proteins. Gene ontology (GO) and pathway analysis showed that these genes were highly associated with IAV infection. The analysis of the PPI network further revealed that the predicted proteins were classified as core proteins within the human protein interaction network. This study may encourage the identification of potential targets for the discovery of more effective anti-influenza drugs. The source codes and datasets are available at https://github.com/HVPPIlab/IVA-Human-PPI/.
Keywords: GO and KEGG; Influenza A virus; Machine learning; Pathogen-host interaction (PHI); Protein-protein interaction (PPI); XGBoost.
© 2025 Li et al.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures







References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
