

Metabolism of hepatic stellate cells in chronic liver diseases: emerging molecular and therapeutic interventions
- PMID: 39897543
- PMCID: PMC11780521
- DOI: 10.7150/thno.106597
Metabolism of hepatic stellate cells in chronic liver diseases: emerging molecular and therapeutic interventions
Abstract
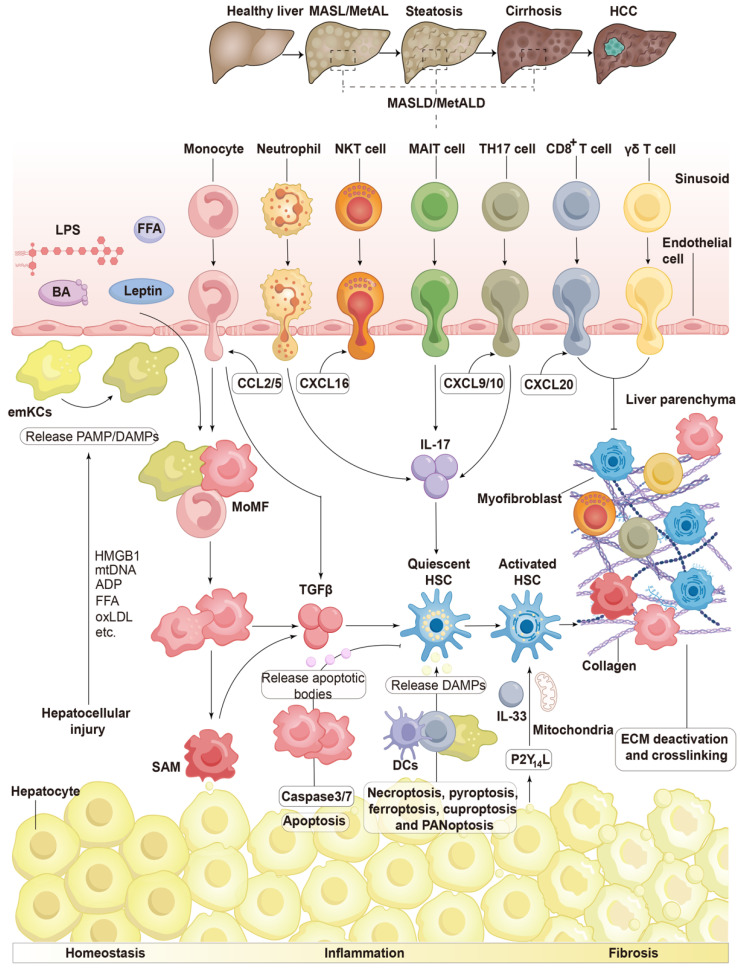
Chronic liver diseases, primarily metabolic dysfunction-associated steatotic liver disease (MASLD), metabolic and metabolic dysfunction-associated alcoholic liver disease (MetALD), and viral hepatitis, can lead to liver fibrosis, cirrhosis, and cancer. Hepatic stellate cell (HSC) activation plays a central role in the development of myofibroblasts and fibrogenesis in chronic liver diseases. However, HSC activation is influenced by the complex microenvironments within the liver, which are largely shaped by the interactions between HSCs and various other cell types. Changes in HSC phenotypes and metabolic mechanisms involve glucose, lipid, and cholesterol metabolism, oxidative stress, activation of the unfolded protein response (UPR), autophagy, ferroptosis, senescence, and nuclear receptors. Clinical interventions targeting these pathways have shown promising results in addressing liver inflammation and fibrosis, as well as in modulating glucose and lipid metabolism and metabolic stress responses. Therefore, a comprehensive understanding of HSC phenotypes and metabolic mechanisms presents opportunities for novel therapeutic approaches aimed at halting or even reversing chronic liver diseases.
Keywords: cellular crosstalk; clinical treatment; hepatic stellate cells; liver diseases.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures




References
-
- Horn P, Tacke F. Metabolic reprogramming in liver fibrosis. Cell Metab. 2024;36:1439–55. - PubMed
-
- Breitkopf-Heinlein K, Martinez-Chantar ML. Targeting hepatic stellate cells to combat liver fibrosis: where do we stand? Gut. 2024;73:1411–3. - PubMed
-
- Sun YD, Zhang H, Li YM, Han JJ. Abnormal metabolism in hepatic stellate cells: Pandora's box of MAFLD related hepatocellular carcinoma. Biochim Biophys Acta Rev Cancer. 2024;1879:189086. - PubMed
-
- Bourebaba N, Marycz K. Hepatic stellate cells role in the course of metabolic disorders development - A molecular overview. Pharmacol Res. 2021;170:105739. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
