

Glutamine missense suppressor transfer RNAs inhibit polyglutamine aggregation
- PMID: 39897579
- PMCID: PMC11787650
- DOI: 10.1016/j.omtn.2024.102442
Glutamine missense suppressor transfer RNAs inhibit polyglutamine aggregation
Abstract
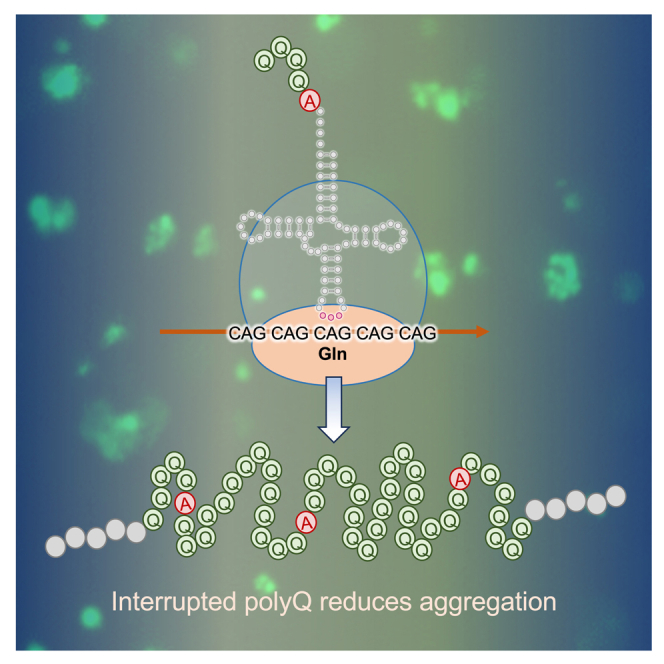
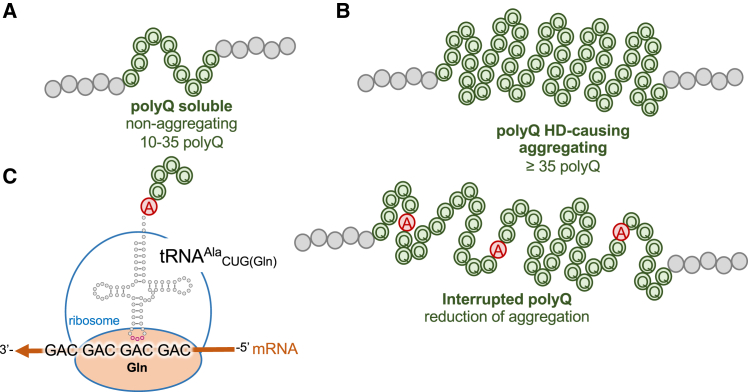
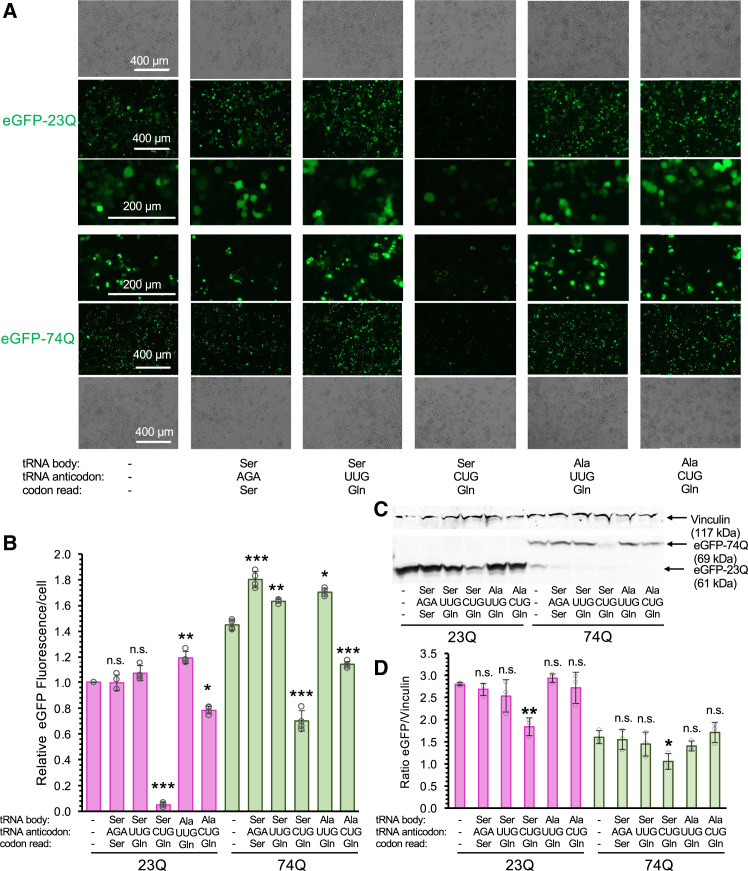
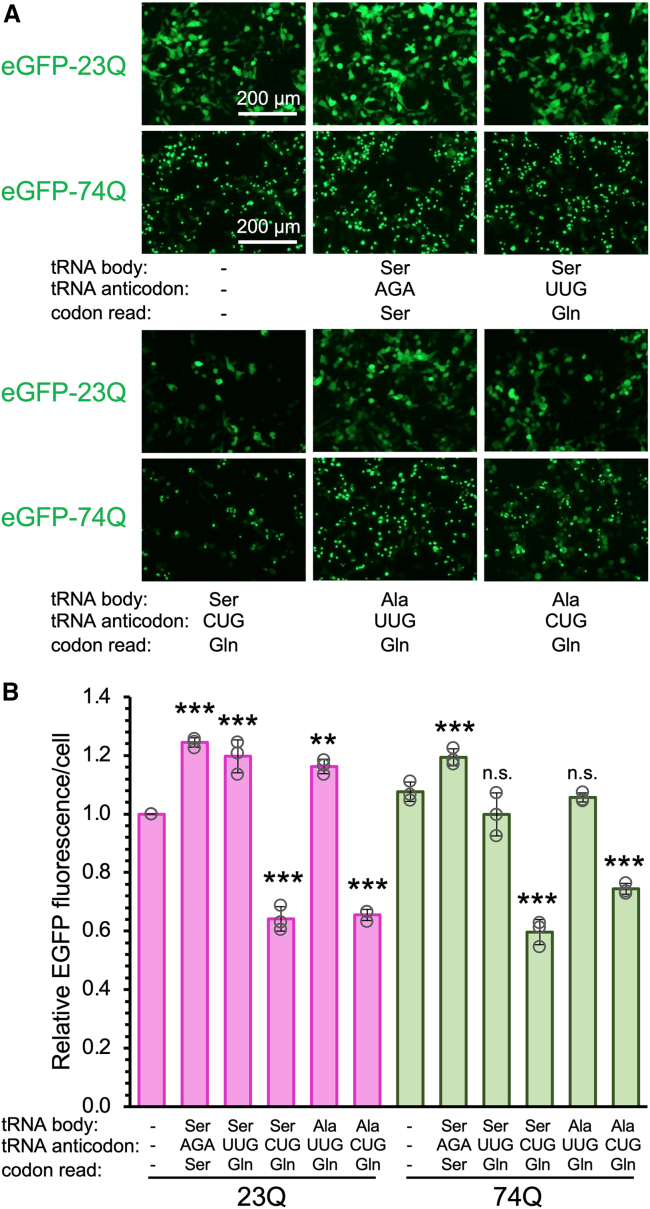
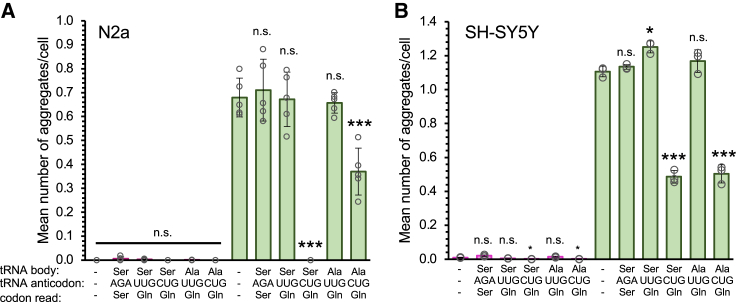
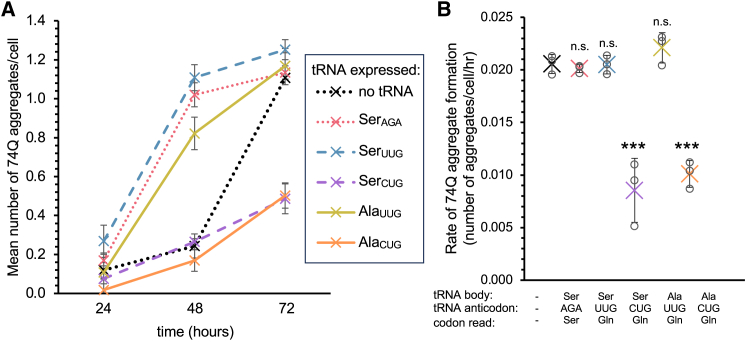
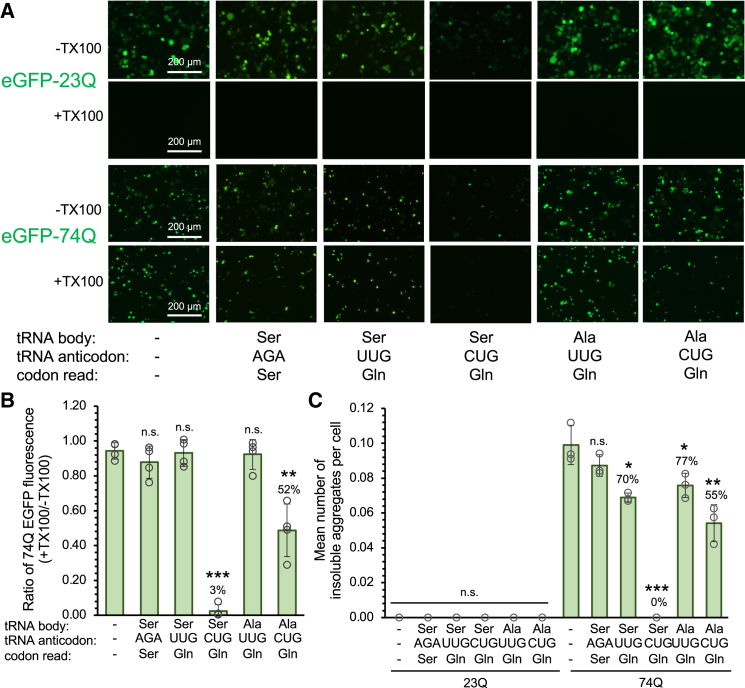
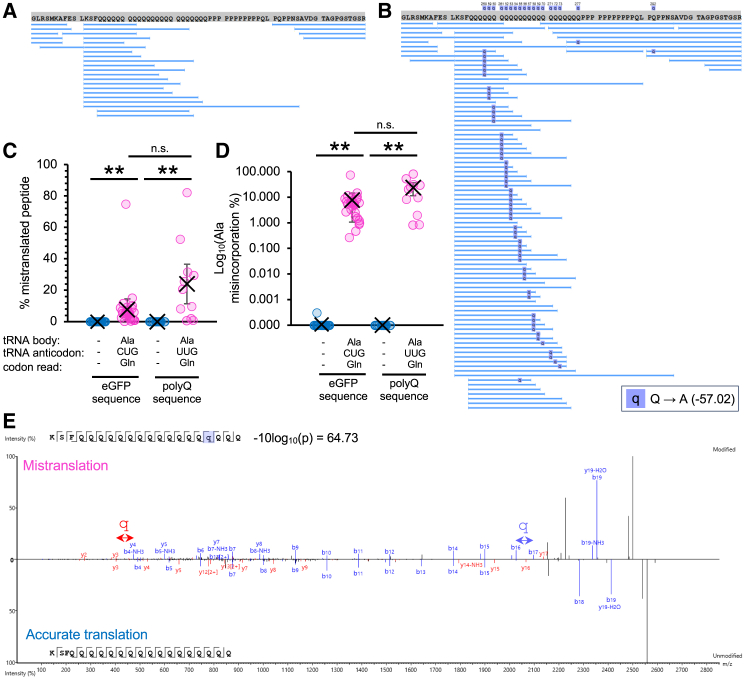
Huntington's disease (HD) is caused by polyglutamine (polyQ) repeat expansions in the huntingtin gene. HD-causative polyQ alleles lead to protein aggregation, which is a prerequisite for disease. Translation fidelity modifies protein aggregation, and several studies suggest that mutating one or two glutamine (Gln) residues in polyQ reduces aggregation. Thus, we hypothesized that missense suppression of Gln codons with other amino acids will reduce polyQ aggregate formation in cells. In neuroblastoma cells, we assessed tRNA variants that misread Gln codons with serine (tRNASer C/UUG) or alanine (tRNAAla C/UUG). The tRNAs with the CUG anticodon were more effective at suppressing the CAG repeats in polyQ, and serine and alanine mis-incorporation had differential impacts on polyQ. The expression of tRNASer CUG reduced polyQ protein production as well as both soluble and insoluble aggregate formation. In contrast, cells expressing tRNAAla CUG selectively decreased insoluble polyQ aggregate formation by 2-fold. Mass spectrometry confirmed Ala mis-incorporation at an average level of ∼20% per Gln codon. Cells expressing the missense suppressor tRNAs showed no cytotoxic effects and no defects in growth or global protein synthesis levels. Our findings demonstrate that tRNA-dependent missense suppression of Gln codons is well tolerated in mammalian cells and significantly reduces polyQ levels and aggregates that cause HD.
Keywords: HD; Huntington's disease; MT: Non-coding RNAs; gene therapy; missense suppression; mistranslation; non-coding RNA; polyQ; polyglutamine; protein aggregation; tRNA therapeutics.
© 2024 The Authors.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Kremer B., Goldberg P., Andrew S.E., Theilmann J., Telenius H., Zeisler J., Squitieri F., Lin B., Bassett A., Almqvist E., et al. A worldwide study of the Huntington's disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 1994;330:1401–1406. doi: 10.1056/nejm199405193302001. - DOI - PubMed
-
- Genetic Modifiers of Huntington’s Disease GeM-HD Consortium Electronic address gusella@helixmghharvardedu. Genetic Modifiers of Huntington’s Disease GeM-HD Consortium CAG Repeat Not Polyglutamine Length Determines Timing of Huntington's Disease Onset. Cell. 2019;178:887–900.e14. doi: 10.1016/j.cell.2019.06.036. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
