

Pathogenic KIAA0586/TALPID3 variants are associated with defects in primary and motile cilia
- PMID: 39898050
- PMCID: PMC11783387
- DOI: 10.1016/j.isci.2024.111670
Pathogenic KIAA0586/TALPID3 variants are associated with defects in primary and motile cilia
Abstract
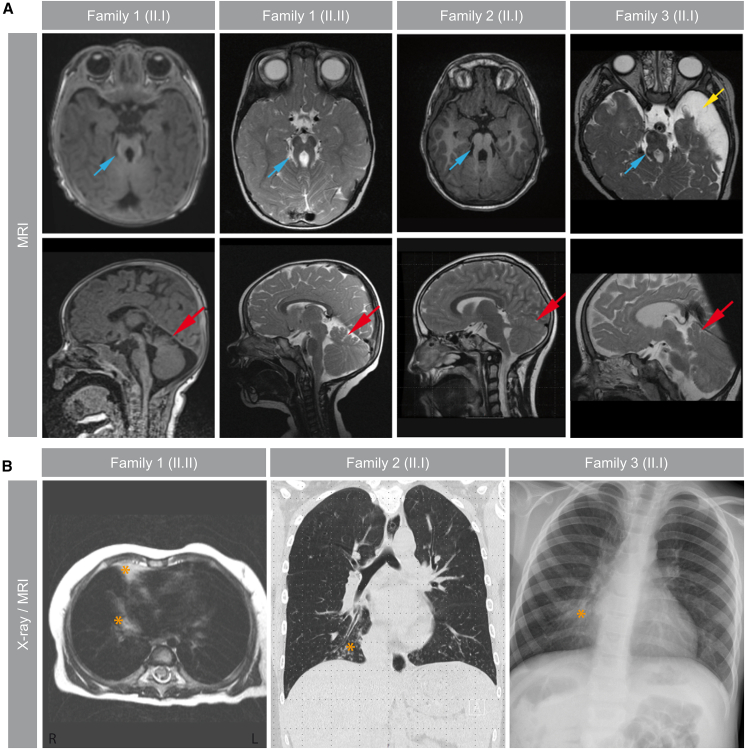
Pathogenic variants in KIAA0586/TALPID3 are associated with the ciliopathy Joubert syndrome (JS). We report individuals with KIAA0586/TALPID3 variants affected by primary and motile cilia defects leading to JS and chronic destructive airway disease. DNA variants were detected in three families by sequencing. In two unrelated families, a deep-intronic variant (KIAA0586/TALPID3:c.3990 + 3186G>A) activated a cryptic exon. We performed histological and functional analyses in native and air-liquid interface (ALI) cultured respiratory cells. Primary cilia lengths were measured in patient-derived fibroblasts. Our data associate KIAA0586/TALPID3 variants with a syndrome combining JS and chronic destructive airway disease, reduced number of motile cilia, disorganized basal body location, and ciliary clearance malfunction. Additionally, patient-derived cell lines showed primary cilia defects. Disease causing KIAA0586/TALPID3 variants, including a deep-intronic sequence variant, were associated with primary and motile cilia defects in JS patients. The combination of JS and respiratory symptoms should be considered indicative for KIAA0586/TALPID3 sequence alterations.
Keywords: Cell biology; Cellular physiology; Human Genetics; Integrative aspects of cell biology.
© 2024 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Maria B.L., Hoang K.B., Tusa R.J., Mancuso A.A., Hamed L.M., Quisling R.G., Hove M.T., Fennell E.B., Booth-Jones M., Ringdahl D.M., et al. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J. Child Neurol. 1997;12:423–430. - PubMed
-
- Alby C., Piquand K., Huber C., Megarbané A., Ichkou A., Legendre M., Pelluard F., Encha-Ravazi F., Abi-Tayeh G., Bessières B., et al. Mutations in KIAA0586 Cause Lethal Ciliopathies Ranging from a Hydrolethalus Phenotype to Short-Rib Polydactyly Syndrome. Am. J. Hum. Genet. 2015;97:311–318. - PMC - PubMed
-
- Parisi M., Glass I. In: GeneReviews® [Internet] Adam M.P., Feldman J., Mirzaa G.M., Pagon R.A., Wallace S.E., Amemiya A., editors. University of Washington; Seattle: 2003. Joubert Syndrome; pp. 1993–2024.https://www.ncbi.nlm.nih.gov/books/NBK1325/ - PubMed
LinkOut - more resources
Full Text Sources
