

A high-resolution bovine mitochondrial co-expression network
- PMID: 39898529
- PMCID: PMC11832118
- DOI: 10.1242/bio.061630
A high-resolution bovine mitochondrial co-expression network
Abstract
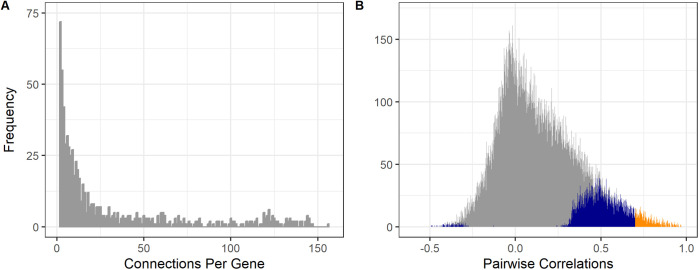
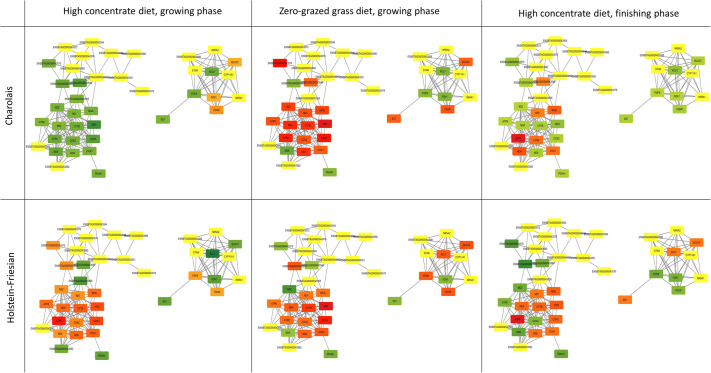
The mitochondrion is a sophisticated, versatile, and dynamic organelle whose function is incompletely understood. Intending to provide a framework for mitochondrial visualisation and interpretation of genome-wide molecular data, we reverse-engineered a co-expression network whose final structure represented mRNA encoding more than half of the entire mitochondrial proteome. We drew upon 723 RNA-seq data sets representing 91 tissues and cell types from 441 individual cattle. A mitochondrial landscape was formed comprising a main network and many smaller sub-networks. One of the discrete sub-networks contains all 13 mRNA (e.g. MT-ND1, MT -CYTB, MT -COX2, MT -ATP8) plus 15/22 tRNA (e.g. MT-TT) encoded by the mt-genome itself, indicating some independent regulation from the nuclear genome with whom it must cooperate. Intriguingly, this mtDNA sub-network also contains a single nuclear-encoded gene, that of PDHA1. PDHA1 encodes a subunit of the pyruvate dehydrogenase complex that governs the conversion of pyruvate to Acetyl CoA. This enzyme is extremely influential, representing the fundamental cellular connection between the ancient, conserved pathway of glycolysis that occurs exclusively in the cytoplasm, and the TCA cycle that occurs within the mitochondrial matrix. To demonstrate the downstream utility of our approach, we overlaid Longissimus dorsi muscle transcriptome data from differentially feed efficient Charolais and Holstein Friesian cattle. This approach highlighted expression patterns sensitive to both breed and diet in a complex manner. An analytic advantage of this approach is that relatively subtle (<2-fold) but coordinated changes that may be overlooked by conventional gene-by-gene significance testing become readily apparent. Finally, intending to understand the transcriptional regulation of mitochondrial function more thoroughly, we engineered a network built with transcription factors in addition to those mRNA encoding mitochondrial proteins. Here, a set of influential nuclear hormone receptors (e.g. PPARA) are enriched among the most highly and/or well-connected TF.
Keywords: Beef cattle; Feed efficiency; Organelle; Partial correlation; RNAseq.
© 2025. Published by The Company of Biologists.
Conflict of interest statement
Competing interests The authors declare no competing or financial interests.
Figures





References
-
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. [cited 2023]. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
