

From bench to bedside: murine models of inherited and sporadic brain arteriovenous malformations
- PMID: 39899215
- PMCID: PMC11790818
- DOI: 10.1007/s10456-024-09953-5
From bench to bedside: murine models of inherited and sporadic brain arteriovenous malformations
Abstract
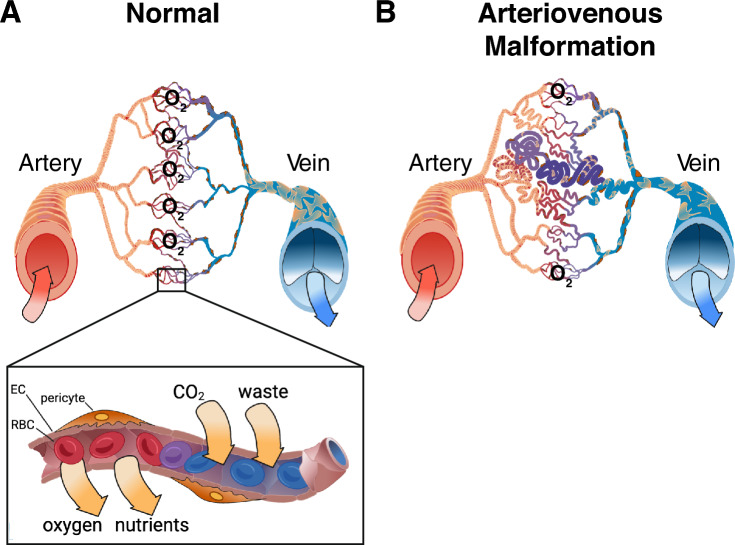
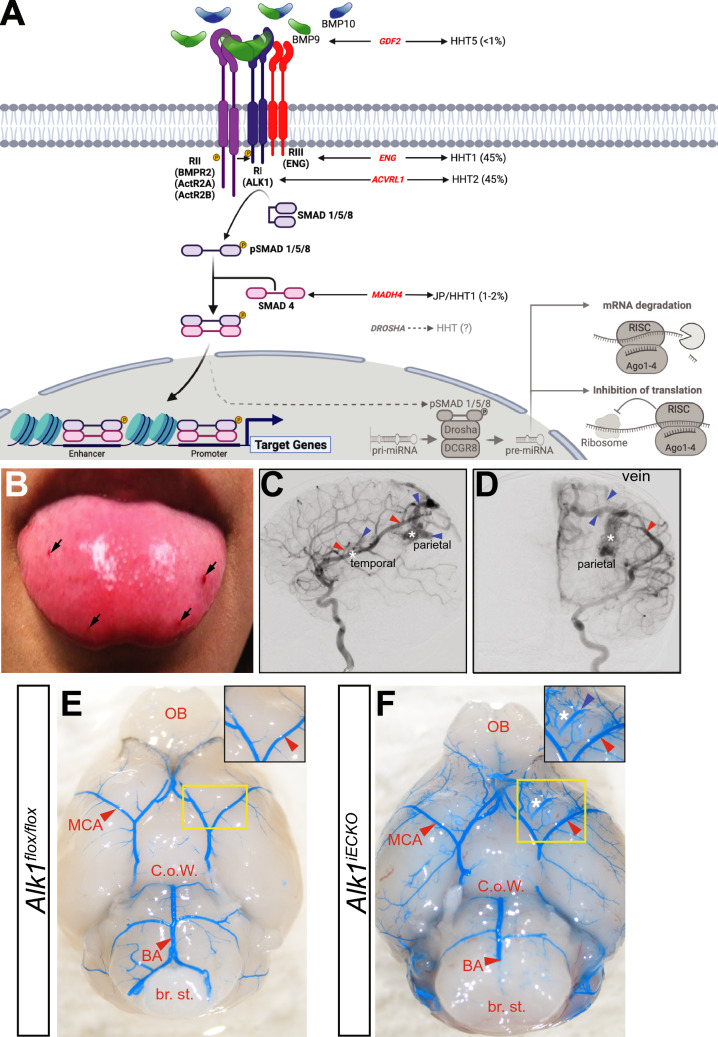
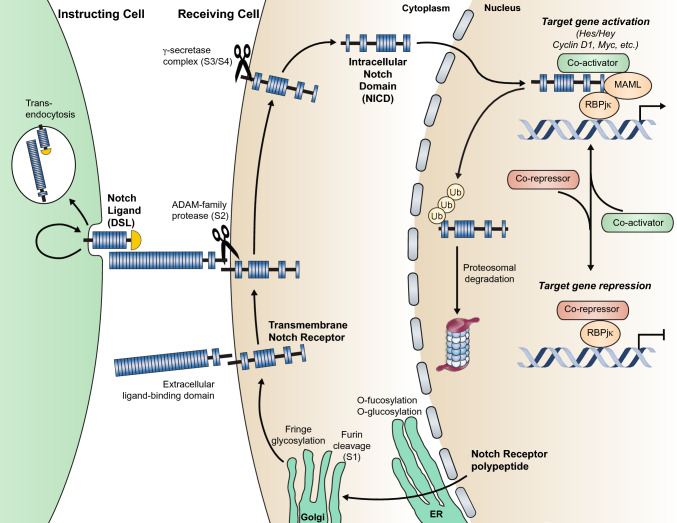
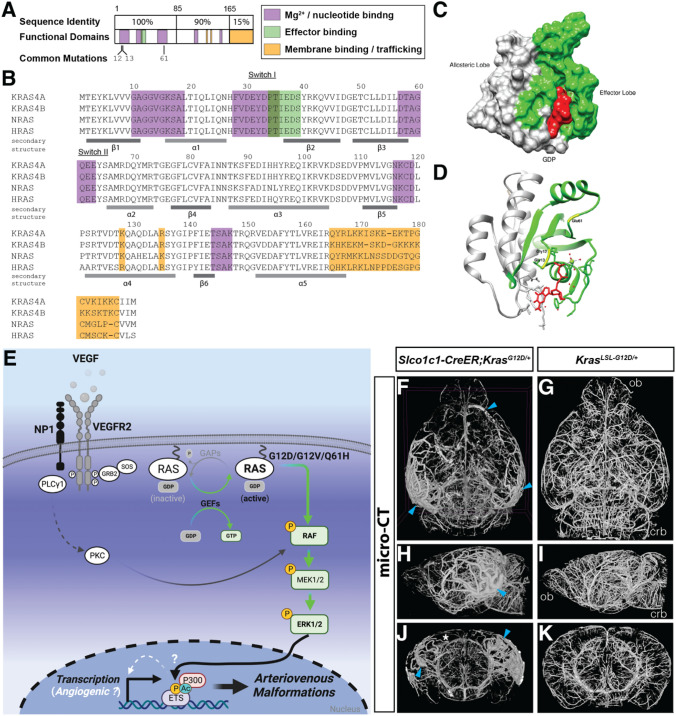
Brain arteriovenous malformations are abnormal vascular structures in which an artery shunts high pressure blood directly to a vein without an intervening capillary bed. These lesions become highly remodeled over time and are prone to rupture. Historically, brain arteriovenous malformations have been challenging to treat, using primarily surgical approaches. Over the past few decades, the genetic causes of these malformations have been uncovered. These can be divided into (1) familial forms, such as loss of function mutations in TGF-β (BMP9/10) components in hereditary hemorrhagic telangiectasia, or (2) sporadic forms, resulting from somatic gain of function mutations in genes involved in the RAS-MAPK signaling pathway. Leveraging these genetic discoveries, preclinical mouse models have been developed to uncover the mechanisms underlying abnormal vessel formation, and thus revealing potential therapeutic targets. Impressively, initial preclinical studies suggest that pharmacological treatments disrupting these aberrant pathways may ameliorate the abnormal pathologic vessel remodeling and inflammatory and hemorrhagic nature of these high-flow vascular anomalies. Intriguingly, these studies also suggest uncontrolled angiogenic signaling may be a major driver in bAVM pathogenesis. This comprehensive review describes the genetics underlying both inherited and sporadic bAVM and details the state of the field regarding murine models of bAVM, highlighting emerging therapeutic targets that may transform our approach to treating these devastating lesions.
Keywords: AVM; BMP; Cerebrovascular; Intracranial hemorrhage; Notch; RAS/MAPK; TGF-β.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interest: The authors declare no competing interests.
Figures
Similar articles
-
Recent Advances in Basic Research for Brain Arteriovenous Malformation.Int J Mol Sci. 2019 Oct 25;20(21):5324. doi: 10.3390/ijms20215324. Int J Mol Sci. 2019. PMID: 31731545 Free PMC article. Review.
-
Somatic mosaicism in the MAPK pathway in sporadic brain arteriovenous malformation and association with phenotype.J Neurosurg. 2021 Jul 2;136(1):148-155. doi: 10.3171/2020.11.JNS202031. Print 2022 Jan 1. J Neurosurg. 2021. PMID: 34214981 Free PMC article.
-
Angiopoietin-2 Inhibition Rescues Arteriovenous Malformation in a Smad4 Hereditary Hemorrhagic Telangiectasia Mouse Model.Circulation. 2019 Apr 23;139(17):2049-2063. doi: 10.1161/CIRCULATIONAHA.118.036952. Circulation. 2019. PMID: 30744395 Free PMC article.
-
Association of common candidate variants with vascular malformations and intracranial hemorrhage in hereditary hemorrhagic telangiectasia.Mol Genet Genomic Med. 2018 May;6(3):350-356. doi: 10.1002/mgg3.377. Epub 2018 Mar 6. Mol Genet Genomic Med. 2018. PMID: 29932521 Free PMC article.
-
Biology of vascular malformations of the brain.Stroke. 2009 Dec;40(12):e694-702. doi: 10.1161/STROKEAHA.109.563692. Epub 2009 Oct 15. Stroke. 2009. PMID: 19834013 Free PMC article. Review.
Cited by
-
Single Cell RNA Sequencing and Spatial Profiling Identify Mechanisms of Neonatal Brain Hemorrhage Development and Resolution.bioRxiv [Preprint]. 2025 Jul 31:2025.07.30.667675. doi: 10.1101/2025.07.30.667675. bioRxiv. 2025. PMID: 40766655 Free PMC article. Preprint.
References
-
- Dalton A, Dobson G, Prasad M, Mukerji N (2018) De novo intracerebral arteriovenous malformations and a review of the theories of their formation. Br J Neurosurg 32(3):305–311. 10.1080/02688697.2018.1478060 - PubMed
-
- Meyer-Heim AD, Boltshauser E (2003) Spontaneous intracranial haemorrhage in children: aetiology, presentation and outcome. Brain Dev 25(6):416–421. 10.1016/s0387-7604(03)00029-9 - PubMed
-
- Celli P, Ferrante L, Palma L, Cavedon G (1984) Cerebral arteriovenous malformations in children: clinical features and outcome of treatment in children and in adults. Surg Neurol 22(1):43–49. 10.1016/0090-3019(84)90227-1 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
