

Bias Dependence of the Transition State of the Hydrogen Evolution Reaction
- PMID: 39900519
- PMCID: PMC11826909
- DOI: 10.1021/jacs.4c18638
Bias Dependence of the Transition State of the Hydrogen Evolution Reaction
Abstract
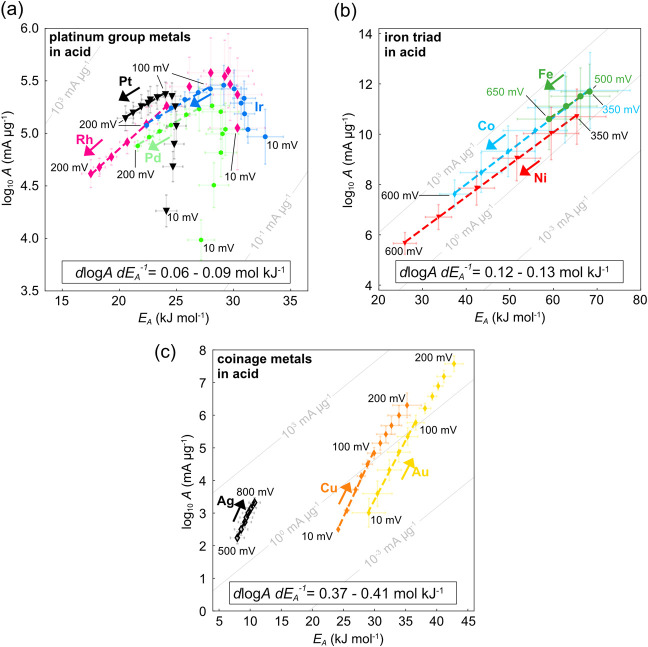
The hydrogen evolution reaction (HER) is one of the most prominent electrocatalytic reactions of green energy transition. However, the kinetics across materials and electrolyte pH and the impact of hydrogen coverage at high current densities remain poorly understood. Here, we study the HER kinetics over a large set of nanoparticle catalysts in industrially relevant acidic and alkaline membrane electrode assemblies that are only operated with pure water humidified gases. We discover distinct kinetic fingerprints between the iron triad (Fe, Ni, Co), coinage (Au, Cu, Ag), and platinum group metals (Ir, Pt, Pd, Rh). Importantly, the applied bias changes not only the activation energy (EA) but also the pre-exponential factor (A). We interpret these changes as entropic changes in the interfacial solvent that differ between acid and base and entropic changes on the surface due to a changing hydrogen coverage. Finally, we observe that anions can induce Butler-Volmer behavior for the coinage metals in acid. Our results provide a new foundation to understand HER kinetics and, more broadly, highlight the pressing need to update common understanding of basic concepts in the field of electrocatalysis.
Conflict of interest statement
The authors declare no competing financial interest.
Figures





References
-
- No̷rskov J. K.; et al. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. 10.1149/1.1856988. - DOI
-
- Ooka H.; Huang J.; Exner K. S. The Sabatier Principle in Electrocatalysis: Basics, Limitations, and Extensions. Front. Energy Res. 2021, 9, 654460 10.3389/fenrg.2021.654460. - DOI
LinkOut - more resources
Full Text Sources
