

A barley pan-transcriptome reveals layers of genotype-dependent transcriptional complexity
- PMID: 39901014
- PMCID: PMC11821519
- DOI: 10.1038/s41588-024-02069-y
A barley pan-transcriptome reveals layers of genotype-dependent transcriptional complexity
Abstract
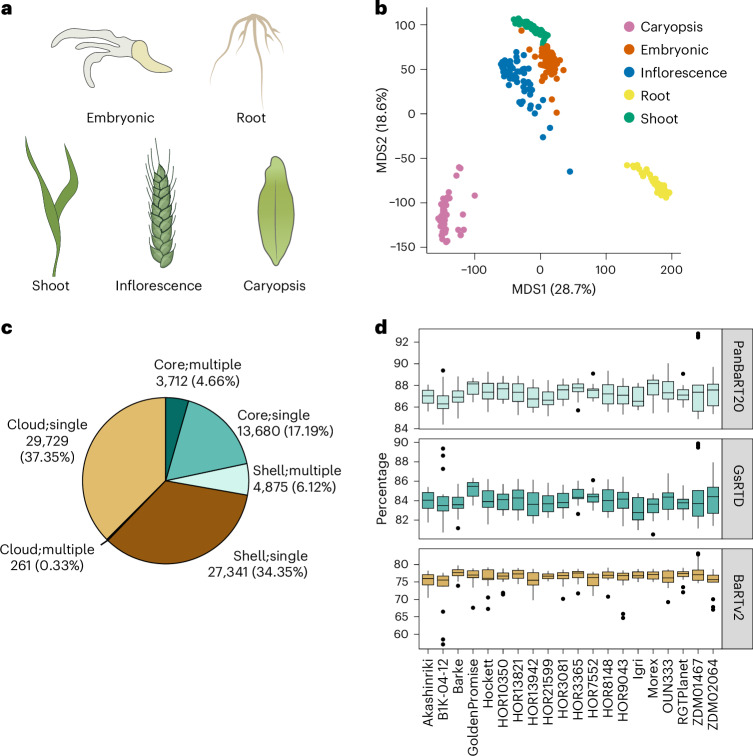
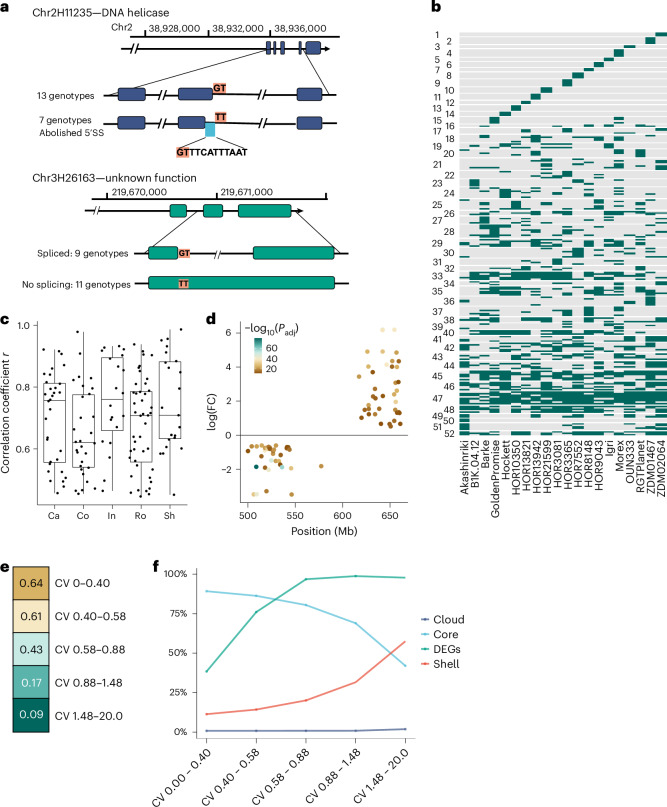
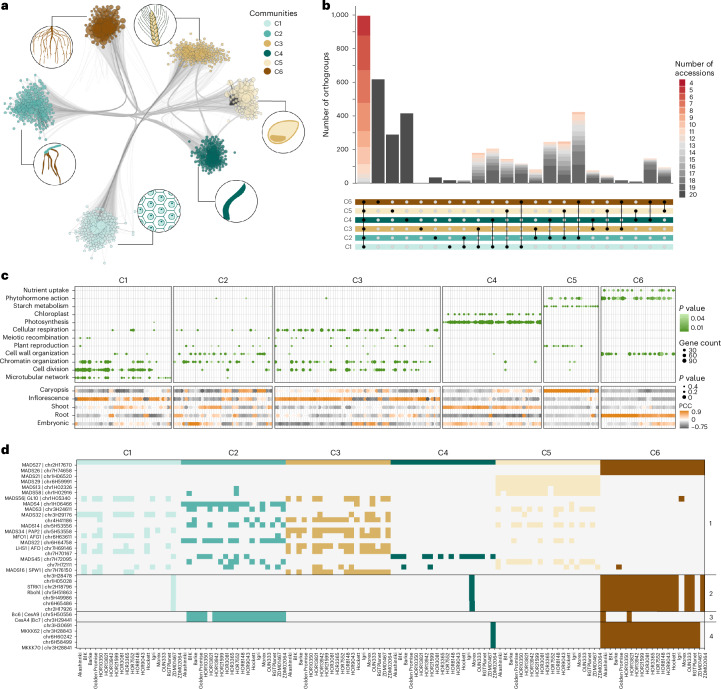
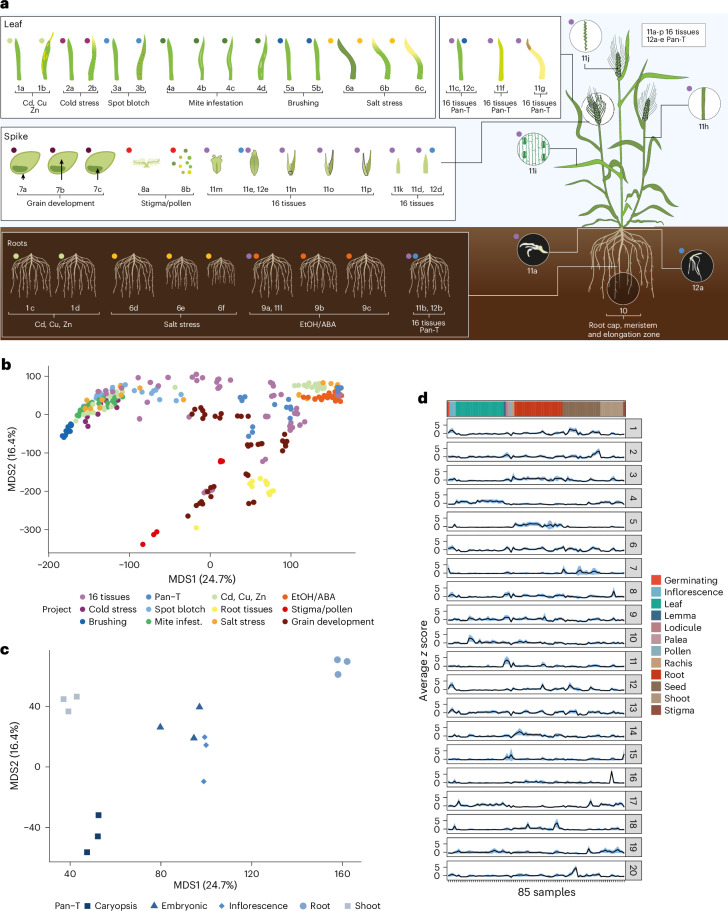
A pan-transcriptome describes the transcriptional and post-transcriptional consequences of genome diversity from multiple individuals within a species. We developed a barley pan-transcriptome using 20 inbred genotypes representing domesticated barley diversity by generating and analyzing short- and long-read RNA-sequencing datasets from multiple tissues. To overcome single reference bias in transcript quantification, we constructed genotype-specific reference transcript datasets (RTDs) and integrated these into a linear pan-genome framework to create a pan-RTD, allowing transcript categorization as core, shell or cloud. Focusing on the core (expressed in all genotypes), we observed significant transcript abundance variation among tissues and between genotypes driven partly by RNA processing, gene copy number, structural rearrangements and conservation of promotor motifs. Network analyses revealed conserved co-expression module::tissue correlations and frequent functional diversification. To complement the pan-transcriptome, we constructed a comprehensive cultivar (cv.) Morex gene-expression atlas and illustrate how these combined datasets can be used to guide biological inquiry.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: K.B.B., C.D., G.B.F., M.E.J., Q.L., M.T.S.N., P.R.P., M.W.R., B.S., N.W.T. and C.V. are employees of the Carlsberg Research Laboratory. The other authors declare no competing interests.
Figures









References
-
- Verstegen, H., Köneke, O., Korzun, V. & von Broock, R. in Biotechnological Approaches to Barley Improvement (eds Kumlehn, J. & Stein, N.) 3–19 (Springer, 2014).
-
- Langridge, P. in The Barley Genome (eds Stein, N. & Muehlbauer, G. J.) 1–10 (Springer, 2018).
-
- Harwood, W. A. in Barley: Methods and Protocols 1–5 (Springer, 2019).
-
- Von Bothmer, R., van Hintum, T., Knüpffer, H. & Sato, K. Diversity in Barley (Hordeum vulgare) Vol. 7 (Elsevier, 2003).
MeSH terms
Grants and funding
- KJHI-B1-2/Rural and Environment Science and Analytical Services Division (Scottish Government's Rural and Environment Science and Analytical Services Division)
- BB/X018636/1/RCUK | Biotechnology and Biological Sciences Research Council (BBSRC)
- BB/S020160/1/RCUK | Biotechnology and Biological Sciences Research Council (BBSRC)
- ERA-CAPS BB/S004610/1/RCUK | Biotechnology and Biological Sciences Research Council (BBSRC)
- UMU1806-002RTX/Grains Research and Development Corporation (Grains Research & Development Corporation)
LinkOut - more resources
Full Text Sources
Research Materials
