

Rescue of common and rare exon 2 skipping variants of the GAA gene using modified U1 snRNA
- PMID: 39905333
- PMCID: PMC11796170
- DOI: 10.1186/s10020-025-01090-z
Rescue of common and rare exon 2 skipping variants of the GAA gene using modified U1 snRNA
Abstract
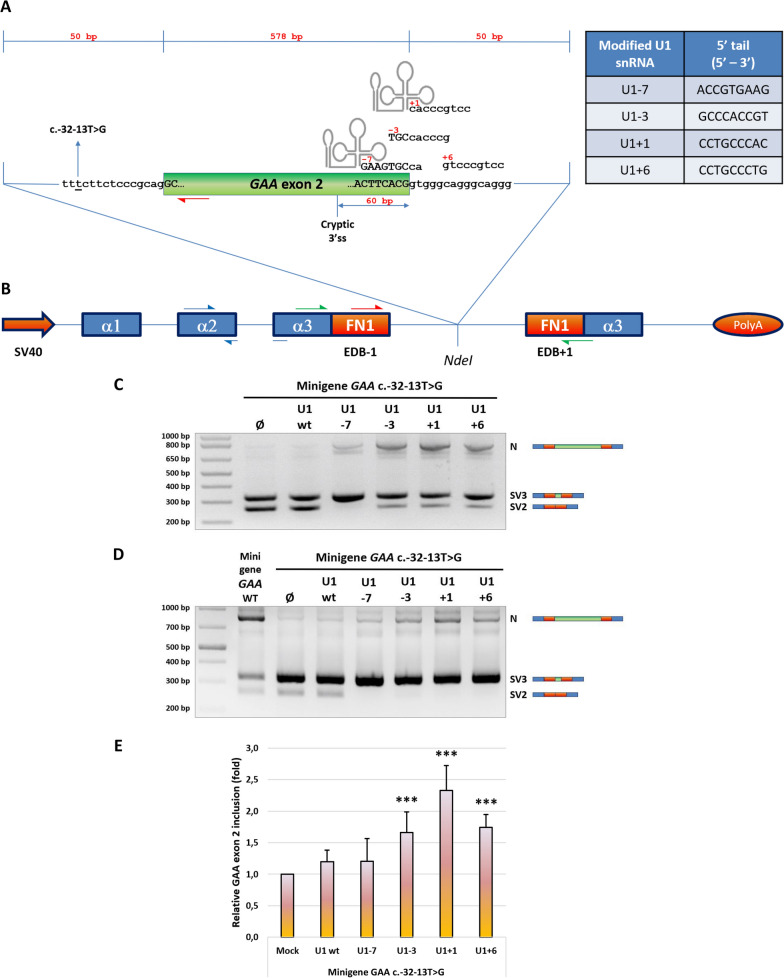
Background: Pompe disease (PD) is an autosomal recessive lysosomal storage disorder caused by the deficient activity of acid alpha glucosidase (GAA) enzyme due to mutations in the GAA gene. As a result, undigested glycogen accumulates within lysosomes causing their dysfunction. From a clinical point of view, the disease can be classified in infantile-onset (IO) and late-onset (LO) forms. The common GAA c.-32-13T>G variant, found in 40-70% of LO-PD alleles, is a leaky splicing mutation interfering with the correct GAA exon 2 recognition by the spliceosome leading to the production of non-functional GAA transcripts. In this study, we used modified, GAA-tailored U1 snRNAs to correct the aberrant splicing determined by the c.-32-13T>G and other GAA exon 2-skipping mutations.
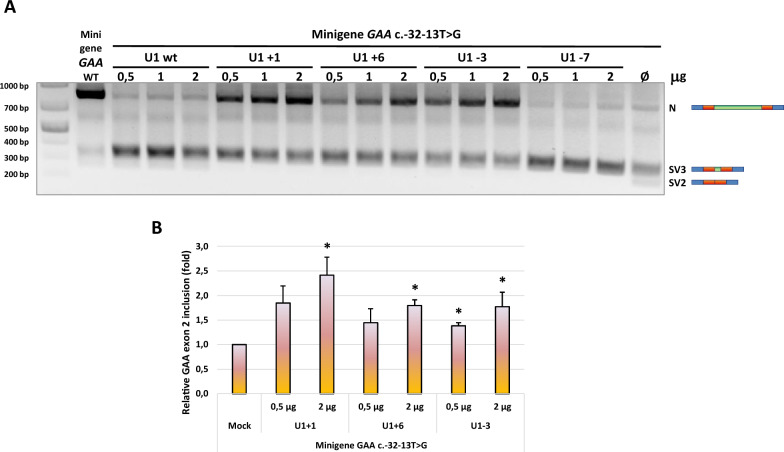
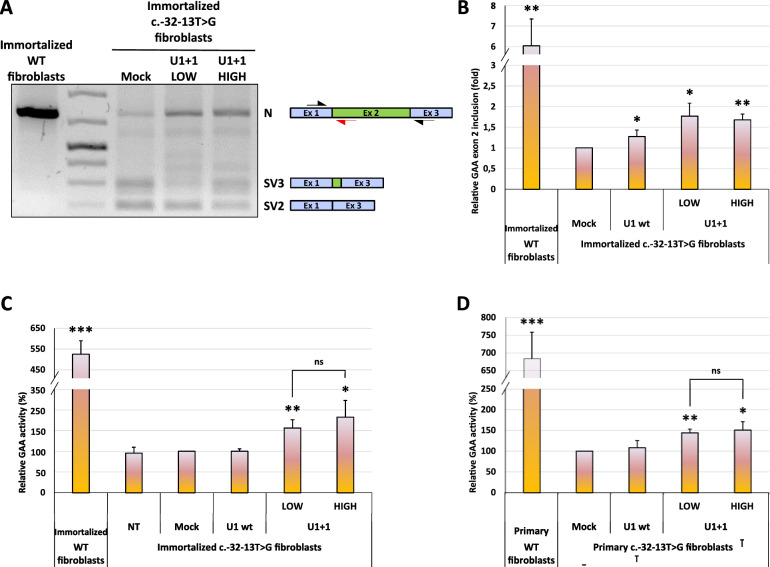
Methods: A set of constructs expressing 5 different engineered U1 snRNAs was generated. A functional splicing assay using a GAA hybrid minigene carrying different variants known to affect GAA exon 2 splicing was used to test the effect of engineered U1 snRNAs on exon 2 inclusion. The effect on endogenously expressed GAA transcript and GAA enzymatic activity was assessed by transfecting patient-derived fibroblasts bearing the common c.-32-13T>G with the best performing modified U1 snRNA.
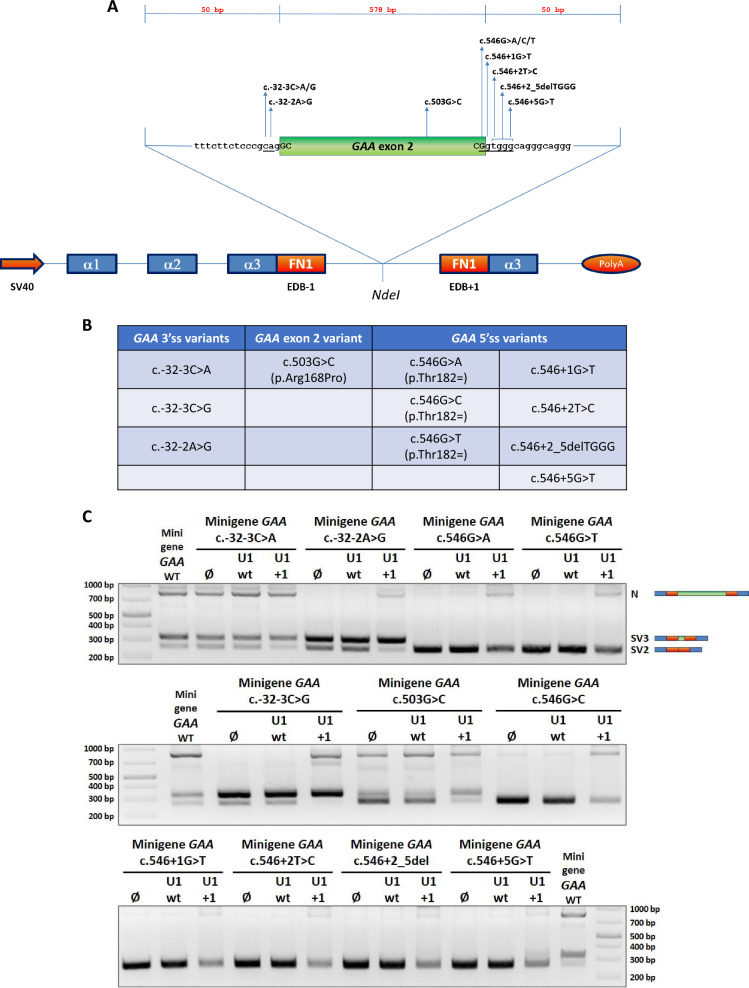
Results: Modified U1-3, U1+1 and U1+6 snRNAs were all able to increase, in a dose-dependent manner, the inclusion of exon 2 within the transcript derived from the GAA minigene harbouring the c.-32-13T>G variant. The U1+1 was the most effective one (2,5 fold increase). Moreover, U1+1 snRNA partially rescued the correct splicing of GAA minigenes harbouring mutations that affect the 3'ss (c.-32-3C>G, c.-32-2A>G) and the 5'ss (c.546G>A, c.546G>C, c.546G>T). Notably, the treatment of patient-derived fibroblasts carrying the c.-32-13T>G mutation with the U1+1 snRNA increased the amount of normal GAA mRNA by 1,8 fold and the GAA enzymatic activity by 70%.
Conclusions: we provide the proof-of-concept for the use of modified GAA-tailored U1 snRNAs, designed to potentiate the recognition of the GAA exon 2 5'ss, as therapeutic tools to correct the aberrant transcripts carrying variants that affect exon 2 splicing, including the common c.-32-13T>G variant.
Keywords: GAA; Glycogenosis type II; Pompe disease; Splicing; U1; c.-32-13T>G.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors have no conflict of interests to declare.
Figures
References
-
- Balestra D, Scalet D, Ferrarese M, Lombardi S, Ziliotto N, Croes CC, Petersen N, Bosma P, Riccardi F, Pagani F, et al. A compensatory U1snRNA partially rescues FAH splicing and protein expression in a splicing-defective mouse model of tyrosinemia type I. Int J Mol Sci. 2020;21(6):2136. 10.3390/ijms21062136. - PMC - PubMed
-
- Baserga SJ, Gesteland SJA, Atkins RF. The RNA World. In: Cold Spring harbor laboratory press. NY; 1993.
-
- Bellotti AS, Andreoli L, Ronchi D, Bresolin N, Comi GP, Corti S. Molecular approaches for the treatment of pompe disease. Mol Neurobiol. 2020;57(2):1259–80. 10.1007/s12035-019-01820-5. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
