

Optimal Transport Based Graph Kernels for Drug Property Prediction
- PMID: 39906265
- PMCID: PMC11793854
- DOI: 10.1109/OJEMB.2024.3480708
Optimal Transport Based Graph Kernels for Drug Property Prediction
Abstract
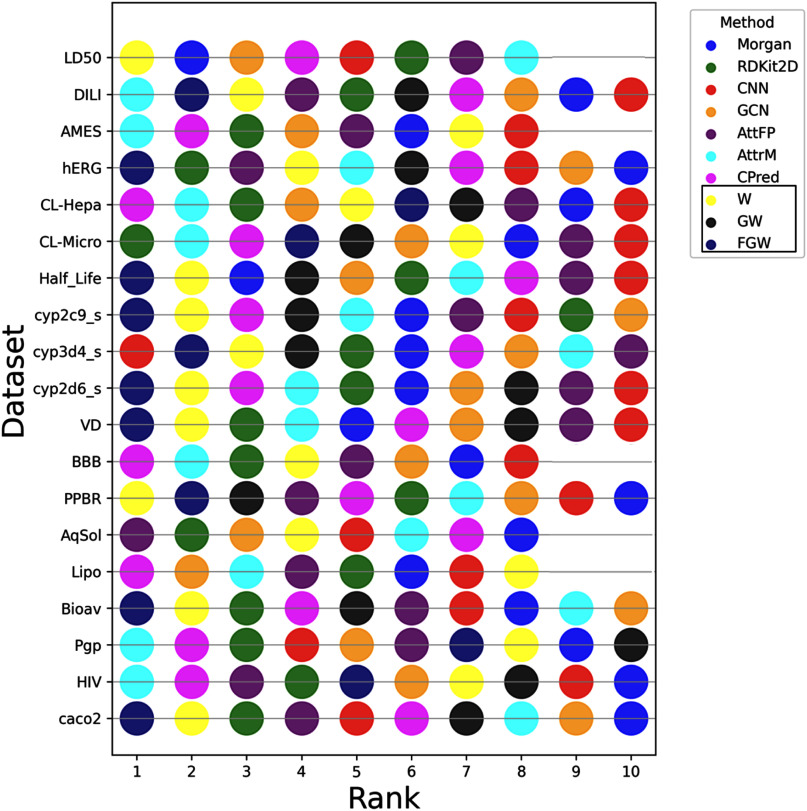
Objective: The development of pharmaceutical agents relies heavily on optimizing their pharmacodynamics, pharmacokinetics, and toxicological properties, collectively known as ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity). Accurate assessment of these properties during the early stages of drug development is challenging due to resource-intensive experimental evaluation and limited comprehensive data availability. To overcome these obstacles, there has been a growing reliance on computational and predictive tools, leveraging recent advancements in machine learning and graph-based methodologies. This study presents an innovative approach that harnesses the power of optimal transport (OT) theory to construct three graph kernels for predicting drug ADMET properties. This approach involves the use of graph matching to create a similarity matrix, which is subsequently integrated into a predictive model. Results: Through extensive evaluations on 19 distinct ADMET datasets, the potential of this methodology becomes evident. The OT-based graph kernels exhibits exceptional performance, outperforming state-of-the-art graph deep learning models in 9 out of 19 datasets, even surpassing the most impactful Graph Neural Network (GNN) that excels in 4 datasets. Furthermore, they are very competitive in 2 additional datasets. Conclusion: Our proposed novel class of OT-based graph kernels not only demonstrates a high degree of effectiveness and competitiveness but also, in contrast to graph neural networks, offers interpretability, adaptability and generalizability across multiple datasets.
Keywords: ADMET properties; Optimal tranpsort; graph kernels; graph matching; wasserstein distance.
© 2024 The Authors.
Conflict of interest statement
The authors assert that they have no conflicts of interest.
Figures
Similar articles
-
Predicting ADMET Properties from Molecule SMILE: A Bottom-Up Approach Using Attention-Based Graph Neural Networks.Pharmaceutics. 2024 Jun 7;16(6):776. doi: 10.3390/pharmaceutics16060776. Pharmaceutics. 2024. PMID: 38931898 Free PMC article.
-
Conformalized Graph Learning for Molecular ADMET Property Prediction and Reliable Uncertainty Quantification.J Chem Inf Model. 2024 Dec 9;64(23):8705-8717. doi: 10.1021/acs.jcim.4c01139. Epub 2024 Nov 21. J Chem Inf Model. 2024. PMID: 39571080
-
FP-GNN: a versatile deep learning architecture for enhanced molecular property prediction.Brief Bioinform. 2022 Nov 19;23(6):bbac408. doi: 10.1093/bib/bbac408. Brief Bioinform. 2022. PMID: 36124766
-
A review on machine learning approaches and trends in drug discovery.Comput Struct Biotechnol J. 2021 Aug 12;19:4538-4558. doi: 10.1016/j.csbj.2021.08.011. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 34471498 Free PMC article. Review.
-
Recent Advances in Toxicity Prediction: Applications of Deep Graph Learning.Chem Res Toxicol. 2023 Aug 21;36(8):1206-1226. doi: 10.1021/acs.chemrestox.2c00384. Epub 2023 Aug 10. Chem Res Toxicol. 2023. PMID: 37562046 Review.
References
-
- Drews J., “Drug discovery: A historical perspective,” Science, vol. 287, no. 5460, pp. 1960–1964, 2000. - PubMed
-
- Ekins S., Ring B. J., Grace J., McRobie-Belle D. J., and Wrighton S. A., “Present and future in vitro approaches for drug metabolism,” J. Pharmacological Toxicological Methods, vol. 44, no. 1, pp. 313–324, 2000. - PubMed
-
- White R. E., “High-throughput screening in drug metabolism and pharmacokinetic support of drug discovery,” Annu. Rev. Pharmacol. Toxicol., vol. 40, no. 1, pp. 133–157, 2000. - PubMed
-
- Kola I. and Landis J., “Can the pharmaceutical industry reduce attrition rates?,” Nat. Rev. Drug Discov., vol. 3, no. 8, pp. 711–716, 2004. - PubMed
-
- Kennedy T., “Managing the drug discovery/development interface,” Drug Discov. Today, vol. 2, no. 10, pp. 436–444, 1997.
